云顶新耀(HKEX 1952.HK),一家专注于创新药及疫苗开发、制造及商业化的生物制药公司,今日公布2024年度业绩报告(截至2024年12月31日)及业务进展。
云顶新耀首席执行官罗永庆表示:“2024年,云顶新耀持续深化‘双轮驱动’战略,已由精准引进海外产品进入到具有出海潜力的自主研发和授权引进的并进模式,专注高价值‘蓝海’领域的同时,致力于研发同类首创或同类最佳的创新疗法。公司全年总收入大幅增长461%,达7.067亿元,超额完成7亿元既定目标;实现高毛利率83%,运营费用占收入比重大幅减少562%,非国际财务准则亏损总额显著收窄25%,现金储备达人民币16亿元,首次实现了年度商业化层面盈利。
三款已上市的核心产品商业化布局高效落地。全球首个对因治疗IgA肾病药物耐赋康®年内实现3.534亿元收入,同比增长1581%。全球首个氟环素类抗菌药物依嘉®,实现收入3.528亿元,同比增长256%,保持强劲的增长势头。自身免疫性疾病领域的同类最佳药物伊曲莫德(VELSIPITY®)已在中国澳门和新加坡获批上市,并通过“港澳药械通”政策成功进入粤港澳大湾区,正式惠及中国内地患者。其新药上市申请已于2024年12月在中国大陆(维适平TM)和中国香港(维长宁TM)获正式受理。
公司持续推进拥有全球权益管线布局,聚焦全球高价值领域,通过自研与全球权益管线实现突破性进展。新一代共价可逆BTK抑制剂EVER001展现出积极的临床数据,全球开发进程稳步推进;公司在mRNA肿瘤和自身免疫疾病治疗领域实现了从基础研究到全球化临床转化的里程碑突破,验证了公司的自主研发能力、AI+mRNA平台的创新优势以及其全球价值。其中,个性化肿瘤治疗性疫苗EVM16已进入临床阶段,并于今年3月完成首例患者给药;通用型现货肿瘤治疗性疫苗EVM14的新药临床试验申请已获美国FDA批准,成为公司首款自研进入全球临床阶段的mRNA肿瘤治疗性疫苗;自体生成CAR-T项目也将于今年完成首个临床前候选药物筛选。云顶新耀拥有位于浙江嘉善的自有商业化规模生产基地,该基地符合全球良好生产规范(GMP)标准,为实现mRNA技术平台的研发、生产以及商业化提供了坚实保障。
展望2025年,我们将继续通过提升商业化运营体系效率与推进自研及全球权益产品差异化管线布局,实现最大化协同效应。公司凭借耐赋康®这款中国唯一获批的IgA肾病疗法,并以其纳入医保作为先发优势,提升市场渗透率从而实现销售放量;积极推进EVER001和AI+mRNA技术平台的创新研发,以全球价值为导向,释放全球合作潜力,加速创新价值的全球化兑现,提升云顶新耀在全球生物制药领域的竞争力。
公司‘双轮驱动’战略已进入全新阶段,云顶新耀将在董事会的指导下,利用已建立的卓越运营的商业化平台自我‘造血’,继续巩固在核心治疗领域的领先地位,持续深化AI+mRNA技术平台的创新研发,朝着2030年成为亚洲领先的全球综合性生物制药公司的愿景稳步迈进,致力于为全球患者提供突破性治疗方案,为股东创造更大回报。”
最新主要产品亮点和预期里程碑
-肾病管线-
耐赋康®(NEFECON®)
2024
2024年3月,本公司授权合作伙伴Calliditas Therapeutics AB(以下称“Calliditas”,已于2024年9月被旭化成集团收购)宣布,基于耐赋康®获得了新适应症的完全批准,美国食品药品监督管理局(FDA)已授予其为期七年的额外孤儿药独占期至2030年12月。
2024年3月,新加坡卫生科学局(HSA)已批准耐赋康®用于治疗具有疾病进展风险的原发性IgA肾病成人患者。新加坡是云顶授权区域内继中国内地和中国澳门后的第三个获得耐赋康®新药上市批准的地区。
2024年4月,Calliditas在2024年国际肾脏病学会(ISN)世界肾脏病大会(WCN 2024)上,公布了耐赋康® NeflgArd III期研究的多项积极结果。此次公布的一项研究结果显示,在2年研究期间(9个月的耐赋康®治疗,停药观察15个月),无论基线尿蛋白肌酐比水平(UPCR)<0.8 g/g 或 ≥0.8 g/g,均能观察到显著的肾小球滤过率(eGFR)获益。另一项研究结果显示,无论基线UPCR如何,耐赋康®组中eGFR降低30%或者进展至肾衰竭的发生率远低于安慰剂组,并且耐赋康®能显著延缓患者eGFR降低30%或进展至肾衰竭的时间。
2024年4月,Calliditas公布了耐赋康® NefIgArd III期研究的全球开放标签扩展(OLE)研究的积极结果。数据显示,在所有IgA肾病患者(IgAN)中,包括在NefIgArd III期研究中接受过耐赋康®治疗的患者,尿蛋白肌酐比水平 (UPCR) 和估算肾小球滤过率 (eGFR) 等终点指标在9个月时的治疗效果与NefIgArd III期研究一致。安全性方面,经过9个月的耐赋康®治疗或在完成NefIgArd III期研究的患者中再次使用耐赋康®治疗后的安全数据与先前报道的安全数据一致。
2024年5月,中国香港卫生署已批准耐赋康®用于治疗有疾病进展风险的原发性IgA肾病成人患者。中国香港是公司授权区域内继中国澳门、中国大陆和新加坡之后的第四个获得耐赋康®新药上市批准的地区。
2024年5月,耐赋康®在中国首张处方成功落地,标志着这款全球首个IgA肾病对因治疗药物开始正式惠及中国大陆的患者,开启国内IgA肾病对因治疗的新篇章。中国是全球原发性肾小球疾病发病率最高的国家之一。此次耐赋康®的首张处方通过互联网医院形式开出,打破时间和地域的限制,为患者及时提供药物,提高患者就医的可及性。
2024年6月,Calliditas在第61届欧洲肾脏协会大会(ERA 2024)上,公布了耐赋康®最新的积极研究结果,数据表明耐赋康®治疗9个月带来的估算肾小球滤过率(eGFR)获益显著优于sparsentan(双效内皮素-血管紧张素受体拮抗剂)持续治疗2年。该研究根据NefIgArd研究的患者特征从PROTECT研究选择匹配人群,使用重复测量混合模型(MMRM)方法分析eGFR的绝对变化,包括基线、3、6、9、12、18和24个月数据、基线eGFR、基线eGFR — 时间交互作用、治疗和治疗 — 时间交互作用。采用MAIC方法评估耐赋康®和sparsentan对9、12和24个月时绝对eGFR变化的影响。研究结果显示,在9、12和24个月时,耐赋康®对eGFR的有利影响均优于sparsentan,且具有统计学和临床显著意义。
2024年7月,中国国家药品监督管理局已正式受理递交的耐赋康®最终临床试验阶段完整数据的补充申请, 这意味着耐赋康®有望成为国内首个且唯一获得中国国家药品监督管理局完全批准的IgA肾病对因治疗药物。
2024年9月,公司宣布耐赋康®作为IgA肾病的唯一对因治疗药物,被纳入《2024版KDIGO IgA肾病和IgA血管炎临床管理实践指南(公开审查版)》,推荐有疾病进展风险的IgA肾病患者进行9个月的耐赋康®治疗(2B)。此次新版指南(草案)在提及目前可用治疗方案的主要优势时指出,耐赋康®是迄今为止唯一被证明可以降低 IgA和IgA免疫复合物水平的治疗方法。
2024年10月,中国台湾地区药政部门(TFDA)已批准耐赋康®用于治疗罹患原发性免疫球蛋白A肾病变(IgAN)且病情有进展风险的成人病人,用以延缓肾功能下降,且无基线蛋白尿水平限制。中国台湾是公司授权区域内继中国澳门、中国大陆、新加坡和中国香港之后的第五个获得耐赋康®新药上市批准的地区。
2024年10月,《肾脏360》(Kidney 360)杂志以“Efficacy and Safety of Nefecon in Patients With Immunoglobulin A Nephropathy From Mainland China: 2-Year NefIgArd Trial Results”《耐赋康®在中国大陆IgA肾病患者中的疗效和安全性:2年NefIgArd试验结果》为题,刊登了耐赋康®在NefIgArd III期研究中完整2年数据的中国亚组数据。文章表示,在2年的治疗和观察期间,中国亚组数据显示,耐赋康®在肾脏保护作用,蛋白尿下降和镜下血尿改善等方面取得了比全球研究中数值上更好的疗效。
2024年10月,耐赋康®NefIgArd研究的最新分析结果在2024美国肾脏病协会肾脏周(ASN Kidney Week 2024)上公布。结果显示,耐赋康®能够在不改变全身IgA反应和总IgA及血清总免疫球蛋白(Ig)水平的情况下,特异性地调节致病性IgA(Gd-IgA1)的产生,进一步验证了耐赋康®是一种耐受性良好、针对性治疗IgA肾病的对因治疗药物。大会上还发布了一项最新真实世界研究数据,结果表明,轻度肾功能受损的IgA肾病患者使用耐赋康®治疗超过9个月可以减少肾功能衰退,保护肾脏,并且耐受性良好。
2024年11月,韩国食品药品安全部(MFDS)已完全批准耐赋康®的新药上市许可申请,用于治疗罹患原发性免疫球蛋白A肾病变(IgAN)的成人患者(尿蛋白≥1.0g/日或尿蛋白与肌酐比(UPCR)≥0.8g/g)。
2024年11月,公司宣布耐赋康®已成功纳入《国家基本医疗保险、工伤保险和生育保险药品目录(2024年)》(“国家医保药品目录”)。新版国家医保药品目录将于2025年1月1日起正式生效。随着耐赋康®此次入保,将有更多中国IgA肾病患者有机会使用这款创新药物,并从中获益。IgA肾病在中国有约500万患者,每年新增确诊患者超过10万人,存在巨大未被满足的需求。
2024年12月,公司宣布耐赋康®于12月16日在香港港怡医院开出中国香港地区首张处方,开启了香港IgA肾病对因治疗的新时代。这也是今年以来继中国内地、新加坡之后,耐赋康®第三个商业化上市的地区。
2025
随着《国家基本医疗保险、工伤保险和生育保险药品目录(2024年)》(“国家医保药品目录”)于2025年1月1日正式实施,耐赋康®开始执行医保新价格,各地IgA肾病患者将陆续享受医保报销。
2025年3月,新加坡卫生科学局已正式批准耐赋康®的补充申请,将其适应症扩展为用于治疗罹患原发性免疫球蛋白A肾病变(IgAN)的成人患者(尿蛋白≥1.0g╱日或尿蛋白与肌酐比(UPCR) ≥0.8g/g)。这意味着耐赋康®成为新加坡完全批准的IgA肾病对因治疗药物。
公司预计将于2025年在韩国和台湾地区商业化上市耐赋康®。
公司预计将在2025年获得中国国家药品监督管理局(NMPA)对耐赋康®的完全批准。
公司预计耐赋康®将被纳入2025年发布的改善全球预后(KDIGO) 2025 指南,并被纳入中国首个 IgA 肾病指南。
EVER001胶囊
EVER001胶囊(又名:XNW1011)是新一代共价可逆的布鲁顿酪氨酸激酶(BTK)抑制剂,正在全球范围内开发用于治疗肾病。BTK是B 细胞受体信号通路的重要组成部分,可调节B淋巴细胞的存活、激活、增殖和分化。应用小分子抑制剂靶向BTK是治疗B细胞淋巴瘤和B细胞调节的自身免疫性疾病的有效选择。中国抗体制药在国内完成的健康受试者I期研究结果表明,EVER001具有高选择性、优异的药代动力学特征、强大靶点结合力和良好的安全性特征,研究结果支持其进一步临床开发。公司拥有在全球开发、生产和商业化EVER001用于治疗肾病的权利。
2024
2024年12月, 公司宣布新一代共价可逆布鲁顿酪氨酸激酶(BTK)抑制剂 EVER001胶囊(又名:XNW1011)在治疗原发性膜性肾病的1b/2a期临床试验阶段性数据取得积极结果。截止到2024年9月13日的数据结果显示,在已完成36周治疗的低剂量组患者中,81.8%(9/11)的患者实现临床缓解,高剂量组中已完成24周治疗的患者已有85.7%(6/7)实现临床缓解。同时,除低剂量组的1例患者外,其他所有完成36周治疗的低剂量组患者,以及所有经24周治疗的高剂量组患者分别在36周和24周都实现了免疫学完全缓解。EVER001总体安全性和耐受性良好。未见在其他共价非可逆BTK抑制剂上观察到的有临床意义的不良事件,如出血、心律失常、严重感染、白细胞减少、血小板减少、严重肝功能损伤等
2025
公司预计在2025年公布1b/2a期临床试验的一年随访数据。
-感染性疾病管线-
依嘉®(依拉环素)
2024
2024年1月,国家卫生健康委临床抗微生物药物敏感性折点研究和标准制定专家委员会评审通过依拉环素的中国临床折点,将使中国的临床医生能够更加合理精准地使用这一新型抗菌药物。
2024年11月,依拉环素(依嘉®)在2024美国感染性疾病周(IDWeek)上公布了多项最新研究结果。一项研究旨在评估依拉环素对碳青霉烯类耐药鲍曼不动杆菌(CRAB)的体外抗菌活性,结果显示了依拉环素体外对CRAB的高度敏感性,也支持在临床微生物实验室中使用MTS和纸片扩散法检测依拉环素的药物敏感性结果一致性和稳定性。另一项研究旨在评估依拉环素对全球范围内临床主要革兰阳性菌和革兰阴性菌的体外活性,共检测了依拉环素(依嘉®)对2018至2022五年期间在亚洲、欧洲、北美洲等多个区域收集的23127株临床病原菌(包括耐药菌株)的敏感性。结果显示,自2018年获批以来,无论地理区域或感染部位如何,依拉环素对所检测的临床相关病原菌都持续保持较高的敏感性,依拉环素上市5年来稳定的体外抗菌活性支持其治疗由革兰阴性和革兰阳性菌引起的复杂性腹腔感染。
2024年11月,国家卫生健康委抗菌药物临床应用与耐药评价专家委员会发起并主办的“依拉环素临床应用综合评价项目”总结会在北京举行,并发布了项目终期报告。依拉环素临床应用综合评价项目自2023年9月23日启动,于2024年11月完成项目数据收集。项目共采集病例3369例,来自全国231家医院的839位医生。终期报告显示,经依拉环素治疗3天时的整体有效率为91.1%,治疗结束时的总治疗有效率为90.1%。
头孢吡肟-他尼硼巴坦
头孢吡肟-他尼硼巴坦是头孢吡肟(一种第四代头孢菌素)和新型β-内酰胺酶抑制剂 (BLI) 他尼硼巴坦的组合,可广泛地覆盖丝氨酸酶和金属 β-内酰胺酶。他尼硼巴坦与头孢吡肟联合使用,正在开发成为一种新的治疗由难治性耐药革兰阴性菌,尤其是碳青霉烯耐药肠杆菌目细菌(CRE)和碳青霉烯耐药或多重耐药铜绿假单胞菌(CRPA/MDR-PA)引起的严重感染的治疗选择。
2024
2024年2月,授权合作伙伴Venatorx Pharmaceuticals宣布《新英格兰医学杂志》(NEJM)发表了头孢吡肟-他尼硼巴坦治疗成人复杂性尿路感染(包括急性肾盂肾炎)患者的3期临床研究CERTAIN-1的积极结果。结果显示头孢吡肟-他尼硼巴坦在治疗复杂性尿路感染(包括急性肾盂肾炎)上优于美罗培南,且安全性与美罗培南相似。
2025
我们预计今年将在中国大陆递交针对复杂性尿路感染(cUTI)的头孢吡肟-他尼硼巴坦的新药上市申请。
-自身免疫性疾病管线-
伊曲莫德 (VELSIPITY®,etrasimod)
2024
2024年2月,授权合作伙伴辉瑞公司宣布欧盟委员会(EC)已授予伊曲莫德(VELSIPITY®)在欧盟的新药上市许可,用于治疗既往对常规疗法或生物制剂反应不足、失去反应或不耐受的16岁及以上中重度活动性溃疡性结肠炎(UC)患者。伊曲莫德是欧盟第一个也是唯一一个获批用于16岁及以上患者的新一代口服治疗溃疡性结肠炎药物。
2024年3月,中国澳门特别行政区药物监督管理局已正式受理伊曲莫德用于治疗中重度活动性溃疡性结肠炎成人患者的新药上市许可申请,并于4月正式获批,澳门成为伊曲莫德在公司亚洲授权区内第一个获得批准的地区。
2024年7月,公司宣布伊曲莫德在治疗中重度活动性溃疡性结肠炎(UC)的亚洲多中心Ⅲ期临床研究的维持期治疗取得积极顶线结果维持期数据证实,在经过40周的维持期治疗后,伊曲莫德组与安慰剂组相比,主要终点和所有关键次要终点均具有显著的临床意义和统计学意义(p<0.0001)的改善,其他包括黏膜愈合、内镜恢复正常等的次要终点也均达到具有显著临床意义和统计学意义(p<0.0001)的改善。维持期治疗显示了伊曲莫德良好的安全性,安全性数据与已知特征一致,没有观察到新的安全性信号。
2024年10月, 得益于“港澳药械通“政策,伊曲莫德正式获得“粤港澳大湾区内地临床急需进口港澳药品批件”批准,并于12月在广东佛山复星禅诚医院开出粤港澳大湾区内地首张处方。伊曲莫德成为云顶新耀第三款商业化新药。
2024年11月,伊曲莫德治疗中重度活动性溃疡性结肠炎(UC)的亚洲多中心Ⅲ期临床研究ES101002的完整诱导期数据在第32届欧洲消化疾病周(UEGW 2024)以口头报告形式公布。ES101002研究是迄今为止在亚洲中重度活动性溃疡性结肠炎患者中完成的最大规模的III期临床研究。诱导期研究结果显示,伊曲莫德治疗组的所有主要和关键次要疗效目标均达到具有统计学意义和临床显著性的改善:与安慰剂组相比的临床缓解率、内镜改善率和临床应答率的治疗差异分别达到 20.4%、28.6%和32.0% ( P值均<0.0001)。接受伊曲莫德治疗的患者在黏膜愈合(P<0.0001)和内镜恢复正常(P=0.0003)方面均达到了具有临床意义和统计学显著性的改善。
2024年12月,中国香港卫生署已正式受理伊曲莫德用于治疗中重度活动性溃疡性结肠炎成人患者的新药上市许可申请。
2024年12月, 伊曲莫德在中国澳门镜湖医院开出首张处方。并被纳入由广东省药品监督管理局、广东省卫生健康委员会发布的广东省粤港澳大湾区内地九市临床急需港澳药品医疗器械目录(2024年)。
2024年12月,中国国家药品监督管理局已正式受理伊曲莫德用于治疗中重度活动性溃疡性结肠炎(UC)患者的新药上市许可申请(NDA)。公司预计会在2026年获得新药上市批准。
2025
2025年2月,伊曲莫德的亚洲多中心Ⅲ期临床研究的完整维持期数据,在第20届欧洲克罗恩病和结肠炎组织大会(ECCO 2025)上以口头报告形式公布。维持期数据显示,伊曲莫德组第40周时达到临床缓解的患者比例显著高于安慰剂组,差异具有显著的临床意义;所有关键次要终点,即维持期第40周内镜改善和临床应答,结果也具有显著的临床意义和统计学意义的改善。其他包括黏膜愈合、内镜恢复正常等的次要终点也均达到具有显著临床意义和统计学意义(p<0.0001)的改善,值得注意的是,51.9%伊曲莫德治疗组患者达到黏膜愈合(定义为中心化阅片内镜子评分≤1[排除易脆]且Geboes指数评分<2.0),而安慰剂组仅有8.8%的患者达到该终点(双侧p值<0.0001)。同时,维持期治疗显示了伊曲莫德良好的安全性,安全性数据与已知特征一致,没有观察到新的安全性信号。
2025年3月,公司宣布启动伊曲莫德位于嘉善工厂的生产建设项目,为伊曲莫德的本地化生产提供支持。该项目总投资为7,000万元,预计正式投产后伊曲莫德的年产能可达5,000万片,计划供应地区覆盖包括中国大陆、中国香港、中国澳门、中国台湾、韩国以及新加坡在内的云顶新耀授权区域。
公司预计将于2025年在中国香港获得伊曲莫德用于治疗中重度活动性溃疡性结肠炎(UC)患者的新药上市批准。
公司预计将于2025年在中国台湾和韩国递交新药上市许可申请。
公司2025年将依托于“港澳药械通”政策,进一步扩大伊曲莫德在大湾区的可及性。
Zetomipzomib(泽托佐米,KZR-616)
Zetomipzomib(泽托佐米,KZR-616)是一款新型、同类首创、选择性免疫蛋白酶体抑制剂,在多种自身免疫性疾病中具有广泛的治疗潜力。公司于2023年9月从Kezar Life Sciences授权引进。公司和Kezar有机会在包括自免性肝炎的自身免疫性疾病领域进行合作。
2024
2024年2月, 中国国家药品监督管理局药品审评中心(CDE)已批准在中国启动zetomipzomib(泽托佐米)治疗活动性狼疮性肾炎(LN)的2b期PALIZADE试验的新药临床试验(IND)申请。
2024年7月, 正在中国推进的zetomipzomib(泽托佐米)治疗活动性狼疮性肾炎(LN)的全球2b期PALIZADE试验已完成中国首例患者给药。
2024年9月,PALIZADE试验在独立数据监测委员会的建议下暂停。10月,Kezar做出了战略性决定,终止PALIZADE试验,并将临床开发重点转向泽托佐米在自身免疫性肝炎(AIH)中的应用。
泽托佐米目前正在进行PORTOLA临床试验,这是一项安慰剂对照、随机、双盲的2a期临床试验,旨在评估该候选药物在AIH患者中的疗效和安全性。AIH是一种罕见的慢性疾病,患者的免疫系统攻击肝脏,导致炎症和组织损伤,严重影响患者的身体健康和生活质量。该研究已完成24名患者的入组,Kezar预计将在2025年上半年公布该临床试验的顶线结果。
-mRNA平台-
云顶新耀正通过具有自主知识产权的mRNA技术平台,多路径开发肿瘤及其他治疗性的mRNA药物,且拥有这些产品的全部知识产权及全部全球权益。公司目前正在研发多个mRNA肿瘤治疗性疫苗药物,包括个性化肿瘤治疗性疫苗(PCV)、肿瘤相关抗原(TAA)肿瘤治疗性疫苗、免疫调节肿瘤治疗性疫苗、自体生成CAR-T产品等,以及新一代脂质纳米颗粒("LNP")递送系统,以增强细胞介导的免疫反应。公司位于中国浙江省嘉善的mRNA生产设施按照全球药品生产质量管理规范("GMP")标准设计,具备临床和商业化规模生产能力。公司对其mRNA治疗性项目拥有完整的全球权益。
2024年,公司积极推进了其mRNA产品管线,EVM16是一款云顶新耀自主研发的新型mRNA个性化肿瘤治疗性疫苗,根据每位患者特有的肿瘤细胞突变,使用自主研发的人工智能算法,预测出具有较高免疫原性潜力的新抗原。mRNA技术平台是我们研发工作的重要组成部分。
2024
2024年2月,公司宣布终止与Providence Therapeutics公司的合作及授权许可协议。协议终止后,公司继续利用该mRNA平台开发预防及治疗性的mRNA自研产品,并拥有这些产品的全部知识产权及全部全球权益。
2024年8月,公司宣布正式启动一款个性化肿瘤疫苗EVM16的研究者发起的临床试验项目(IIT)EVM16CX01。该研究由北京大学肿瘤医院和复旦大学附属肿瘤医院发起,用于评估EVM16注射液单药及联合PD-1 抗体治疗在晚期或复发实体瘤受试者的安全性、耐受性、免疫原性和初步疗效的剂量递增及扩展研究。这是EVM16开展的首次人体试验。
2025
2025年3月,公司宣布自主研发的首款新型mRNA个性化肿瘤治疗性疫苗EVM16已在北京大学肿瘤医院顺利完成首例患者给药,标志着此项临床试验项目的里程碑进展。
2025年3月,公司宣布其通用型的现货肿瘤治疗性疫苗EVM14注射液的新药临床试验申请(“IND”)获美国食品药品监督监督管理局(“FDA”)的批准。EVM14注射液是基于云顶新耀自主知识产权的mRNA技术平台研发。作为公司首个获得FDA IND批准的自主研发新药,EVM14标志着公司在mRNA肿瘤领域的创新实力获得国际权威机构的认可,是公司自主研发历程中的重要里程碑。
公司预计将于2025年在中国递交EVM14-肿瘤相关抗原(TAA)疫苗的新药临床试验申请。
产品管线
本公司已在肾脏、抗感染和自身免疫性疾病领域建立了强大的产品管线,包括疾病首创或同类最佳资产。这些项目涵盖短期、中期及长期机会,预计将为公司带来显著的收入增长,并为股东创造价值。
下表概列截至最后实际可行日期,我们的主要产品管线及各候选药物及疫苗的开发状况:
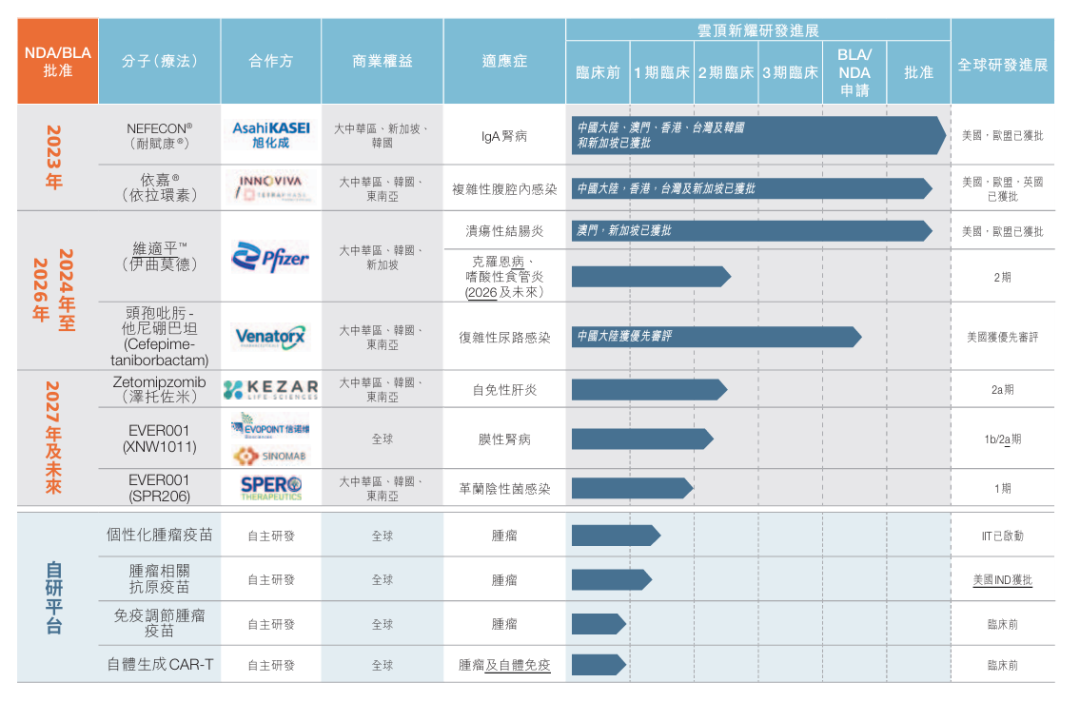
管线展望
公司拥有全球权益并有潜力成为同类领先(best-in-class)的项目预计将在2025年取得突破性进展,包括关键数据读出。2024年,EVER001(一种新型BTK抑制剂,开发的第一个适应症为原发性膜性肾病)的1b/2a期临床试验中获得积极的阶段性数据结果。我们将在2025年持续推进该临床试验,并预计在下半年公布一年的随访数据。
在自主研发管线方面,我们的个性化肿瘤疫苗项目已取得重要进展:EVM16于2025年3月在北京大学肿瘤医院顺利完成首例患者给药,表明云顶新耀具有自主知识产权的肿瘤新抗原人工智能(AI)算法系统和经过临床验证的mRNA技术平台已成功进入人体试验阶段。我们预计将在今年获得该肿瘤疫苗的安全性和免疫原性初步数据。公司的现货型肿瘤相关抗原疫苗的新药临床试验申请已获美国FDA的批准,同时计划于今年上半年向中国药监局递交IND申请,这是公司首次获得美国FDA的IND批准,同时也是公司自主研发管线的首次中美两地同步申报,彰显了我们在创新药物开发领域的重大突破。
关于我们的临床后期项目,公司计划于2025年在中国大陆递交头孢吡肟-他尼硼巴坦用于治疗复杂性尿路感染的新药上市许可申请。我们还计划将在韩国和中国台湾递交伊曲莫德的新药上市许可申请,该产品预计下半年在香港获得新药上市批准。
商业化进展
2024年,随着耐赋康®、依嘉®和伊曲莫德三款核心产品的成功上市及市场拓展,我们的商业化能力实现了显著提升,业务规模持续扩大。
在肾病治疗领域,我们迎来了重大里程碑——疾病首创的创新药物耐赋康®于2024年5月正式在中国大陆上市。为满足国内约500万IgA肾病患者的迫切治疗需求,我们制定了创新的市场策略:在打开传统医院渠道、开展专业学术推广的同时,率先布局互联网医疗平台,开创性地构建了线上线下协同的处方模式。截至2024年底,凭借150人规模的专业肾病销售团队,我们已成功覆盖全国600-700余家核心医院,触达60%以上的目标患者群体。尤为重要的是,在全体员工的不懈努力下,耐赋康®成功纳入《国家基本医疗保险、工伤保险和生育保险药品目录(2024年)》(“国家医保药品目录”),该目录于2025年1月1日起正式实施。我们预计,医保报销政策的落地将大幅提升耐赋康®的可负担性和可及性,有力推动产品在目标患者群体中的快速渗透,进一步夯实耐赋康®作为IgA肾病的一线基础用药地位。
耐赋康®在2024年的另一项重要突破是其作为IgA肾病的唯一对因治疗药物被纳入《2024 KDIGO IgA肾病和IgA血管炎临床管理实践指南(公开审查版)》。指南草案指出耐赋康®是迄今为止唯一被证明可以降低致病性IgA和IgA免疫复合物水平的治疗方法,建议对有疾病进展风险的IgA肾病患者进行9个月的耐赋康®治疗(2B),并提出多数患者可能需要重复9个月治疗周期或降低剂量的维持方案,以在降低蛋白尿及保护肾功能方面产生持续的临床效果。合作伙伴Calliditas的全球开放标签扩展研究亦证实了重复治疗的安全性和疗效,该研究显示,接受耐赋康®第二个治疗疗程的IgA肾病患者在肾功能保护和蛋白尿获益上与首次治疗患者的疗效相当,且耐受性良好。

此外,我们很高兴推出了首个自身免疫性疾病治疗药物伊曲莫德,该药物于去年4月在澳门获批,5月在新加坡获批,并成功在这些地区商业化上市。得益于“港澳药械通”政策,伊曲莫德获得了在广东省九个城市的先行使用资格,并于12月在佛山复星禅诚医院开出了首张处方。截至目前,伊曲莫德已在广东省的五家指定医疗机构(中山大学附属第一医院、佛山复星禅诚医院、南方医科大学深圳医院和广州和睦家医院和深圳前海蛇口自贸区医院)可及,以满足对先进创新药物有迫切需求的中国大陆溃疡性结肠炎患者的治疗需求。
2025年,我们将依托去年打下的坚实商业化基础,全面推进各个疾病领域的业务发展。随着耐赋康®成功纳入国家医保药品目录,患者可负担性和可及性显著提升,有望为公司带来强劲的销售增长动力。我们计划第一季度实现70%核心市场的医保覆盖,我们计划今年第一季度快速推进目标医院完成医保落地,包括通过医院准入或双通道药房;同时,通过200名左右销售代表组成的专业肾科团队,将目标医院扩展至约800家,覆盖超过70%的耐赋康®潜力市场。今年上半年,我们还预计耐赋康®将被正式纳入2025年修订版《改善全球肾脏病预后组织(KDIGO)IgA肾病和IgA血管炎临床管理实践指南》,并被纳入中国首部IgA肾病诊疗指南,获得一线对因治疗药物的权威推荐,这将为临床医生提供重要的用药依据。在深耕中国大陆市场、充分把握耐赋康®医保定价带来的先发优势的同时,我们还将积极拓展其它亚洲市场,包括中国台湾和韩国等地区,让更多IgA肾病患者能够尽快获益于这一突破性疾病首创药物。
在抗感染产品管线中,我们仍将重点拓展依嘉®在核心医疗机构的市场潜力,通过系统性学术推广和临床价值传递,提升医生对依嘉®治疗优势的认知度,从而实现高潜力医院处方量的稳步增长。同时,我们将着力加速推动该药物在重症医学科(ICU)、呼吸科和血液科等科室中治疗时机前移,利用其抗菌活性强、抗菌谱广、组织浓度高和安全性佳等优势强化依嘉®在多重耐药菌感染经验性治疗中的基石地位。在渠道拓展方面,除内部销售团队外,我们还将通过深化CSO模式落地,进一步扩大依嘉®的市场覆盖和临床应用,拯救更多的重症患者。
伊曲莫德是我们最新推出的自身免疫性疾病产品,公司将在2025年全力配合中国药监局的新药上市许可申请审评工作,该申请预计将于2026年获得批准。在此期间,我们也将积极推动该药物通过港澳药械通政策在广东省更多的指定医疗机构中可及,以造福更多的中国大陆患者。同时,我们计划在大湾区开展伊曲莫德的真实世界研究,为医生提供更多循证医学依据和临床治疗指导。
新药发现
2024年是公司mRNA技术平台取得显著进展的一年,我们在中国两家顶级肿瘤医院启动了个性化肿瘤疫苗EVM16的研究者发起项目的首次人体试验。EVM16是一款云顶新耀自研、AI算法驱动识别肿瘤新抗原的新型mRNA个性化肿瘤治疗性疫苗,可根据每位患者特有的肿瘤细胞突变,使用自主研发且具备自我迭代能力的EVER-NEO-1“妙算”肿瘤新抗原人工智能AI算法系统,识别出具有较高免疫原性的肿瘤新抗原,并设计出编码数十种肿瘤新抗原的mRNA治疗性疫苗。EVM16通过LNP递送系统在体内进行高效的抗原呈递,激活患者自身的新抗原特异性T细胞免疫,进而达到杀伤肿瘤细胞和治疗癌症的目的。
在此前的临床前研究中,EVM16在多种小鼠模型中都激发出了强烈的新抗原特异性T细胞免疫反应,在小鼠黑色素瘤B16F10模型中实现了显著的抑制肿瘤生长的药效。云顶新耀自研的EVER-NEO-1“妙算”肿瘤新抗原人工智能AI算法系统不仅能识别出绝大多数已被报道的肿瘤新抗原,还识别出了多个之前未报道的肿瘤新抗原,并且在多个独立验证研究中展现出与行业领先算法相当或更优的新抗原免疫原性预测能力。临床前数据还证明了EVM16与PD-1抗体联用后具有对T细胞激活具有协同效果,支持个性化肿瘤疫苗与免疫检查点抑制剂在临床中的联用。在临床前安全性评估试验中,EVM16也展现了良好的安全性。这些结果综合说明,EVM16注射液免疫原性强、安全性良好,和免疫检查点抑制剂联用有望给肿瘤患者带来更多临床获益。
我们期待2025年将是充满机遇的一年,个性化肿瘤疫苗EVM16有望在年内获得初步人体数据结果。此外,我们已向美国FDA递交了通用型的现货肿瘤相关抗原疫苗EVM14的新药临床试验申请并获得了批准,作为公司首个获得FDA IND批准的自主研发新药,EVM14标志着公司在mRNA肿瘤领域的创新实力获得国际权威机构的认可,是公司自主研发历程中的重要里程碑。该候选产品有潜力用于治疗多种实体瘤适应症,如非小细胞肺癌(NSCLC)和头颈癌等。在临床前试验中,EVM14在小鼠中诱导了剂量依赖性的抗原特异性免疫应答,并在多个小鼠同源肿瘤模型中显著地抑制了肿瘤生长。同时,我们的自体生成CAR-T项目正积极向临床前候选分子选择阶段推进,有望为未来的全球合作创造机会。
业务拓展
基于我们拥有全球权益产品EVER001所展现的卓越临床数据,公司正积极推进对外授权合作,计划寻求全球合作伙伴,希望能够通过整合全球研发资源和专业优势,最大化该项目和其它早期资产的经济价值。在授权引进战略上,我们持续聚焦于肾病、自身免疫性疾病及抗感染等具有高临床价值且竞争格局良好的治疗领域,重点引进具有"同类首创"或"同类最佳"潜力的创新资产。在中国市场,基于我们已建立的高效精干的商业化模式,我们将着眼于引进商业化阶段产品,以利用我们经验丰富且高效的销售团队,实现协同效应最大化,提升运营效率。同时,我们也将战略性评估具有全球权益的早期资产,可通过高效的临床开发实现概念验证,为股东创造显著价值回报。
财务亮点
国际财务报告准则数字:
截至2024年12月31日止年度的收益大幅增加人民币580.7百万元或461%至人民币706.7百万元,而截至2023年12月31日止年度为人民币125.9百万元。收益增加主要由于依嘉®销售额强劲增长及耐赋康®于中国大陆成功上市所致。此外,在中国大陆以外的市场,依嘉®在香港及新加坡的销售额继续上升,耐赋康®于香港及新加坡成功上市,而VELSIPITY®首次于澳门上市,并通过“港澳药械通”政策于广东省上市。
毛利率由截至2023年12月31日止年度的72.7%增加至截至2024年12月31日止年度的74.6%。剔除无形资产摊销后,毛利率由2023年的79.9%增加至2024年的82.9%。有关改善主要由于耐赋康®的商业上市及优化产品成本。
截至2024年12月31日止年度的研发开支为人民币528.0百万元,较截至2023年12月31日止年度的人民币540.1百万元略有减少。本公司继续致力于对不同产品线的策略性研发投资,以支持长期的可持续增长。
一般及行政开支由截至2023年12月31日止年度的人民币165.2百万元增加人民币84.9百万元至截至2024年12月31日止年度的人民币250.1百万元。该增加主要由于薪酬开支及专业服务开支增加,以支持本公司的业务扩展及持续管线增长
分销及销售开支由截至2023年12月31日止年度的人民币231.4百万元增加人民币276.7百万元至截至2024年12月31日止年度的人民币508.1百万元,主要由于扩大商业团队及额外商业活动,此有助于新产品的成功上市并促进现有产品销售的增长。随着我们继续搭建更有效率及更聚焦的商业化模式,销售费用率下降111.9%,我们的商业运营效率有所提升。
经营开支总额(包括一般及行政开支、研发开支以及分销及销售开支)占销售额的比率下降561.8%,表明运营效率有所提升。
年内亏损净额由截至2023年12月31日止年度的人民币844.5百万元增加人民币196.9百万元至截至2024年12月31日止年度的人民币1,041.4百万元。该增加主要由于2024年上半年与mRNA COVID-19疫苗有关的无形资产的一次性、非经常性减值亏损。
剔除无形资产减值亏损后,亏损净额由截至2023年12月31日止年度的人民币792.5百万元收窄人民币107.5百万元至截至2024年12月31日止年度的人民币685.0百万元。此主要由于产品销售强劲及运营效率提升所致。
截至2024年12月31日,现金及现金等价物以及银行存款为人民币1,603.3百万元。
非国际财务报告准则数字:
年内经调整亏损由截至2023年12月31日止年度的人民币713.6百万元收窄人民币176.1百万元至截至2024年12月31日止年度的人民币537.6百万元,主要是剔除无形资产减值的一次性及非经常性亏损、以股份为基础的薪酬的非现金开支及无形资产摊销。
关于云顶新耀
云顶新耀是一家专注于创新药和疫苗研发、临床开发、制造及商业化的生物制药公司,致力于满足亚洲市场尚未满足的医疗需求。云顶新耀的管理团队在中国及全球领先制药企业从事过高质量研发、临床开发、药政事务、化学制造与控制(CMC)、业务发展和商业化运营,拥有深厚的专长和丰富的经验。云顶新耀已打造多款疾病首创或者同类最佳的药物组合。公司的治疗领域包括肾科疾病、感染性和传染性疾病、自身免疫性疾病。