• 结束了以往先进治疗药品(ATMP)概念模糊、归类分散的局面,而且清晰的分类标准能够为药品研发立项、技术路线选择提供明确指引;
• 美国、欧盟均已单独设立ATMP审评机构;
• 产品一旦被认定为ATMP,企业更关心的是能否享受ATMP加速审评路径;
• 818号文为干细胞、CAR-T、体内/体外基因编辑等尚未达到药品注册标准的早期ATMP,明确了从非临床研究、临床研究到转化应用的完整路径。
2025年,国家药监局首次以法规形式明确先进治疗药品(ATMP)的定义及细胞治疗、基因治疗、其他三大类核心分类。回顾过去5年间,药监部门发布26项ATMP技术指南,构建起覆盖CMC、非临床、临床药理、临床及GMP、变更管理的全生命周期监管技术矩阵。可以说,当前我国ATMP监管正经历从概念模糊到规则引领的关键转型。
监管政策迭代的背景下,是我国ATMP产业的快速发展。迄今国内已有10个ATMP疗法上市,包括8个CAR-T、1个干细胞疗法、1个基因疗法;新发布的创新药商保目录中纳入了5个CAR-T药物。同时我国ATMP发展也在赶超国际,据统计,我国细胞治疗临床试验数量及申报产品数量位居全球第二位,仅次于美国。
如何让监管紧跟并引领ATMP创新发展需求?接受研发客采访的业内专家认为,接下来需针对不同亚类产品细化审评要点、制定指南,并建议尽快成立专门审评部门以提升审评专业度。他们还提出,待分类完全明晰后,结合临床需求与数据综合评估,再审慎探索建立ATMP专属的特殊审评通道。
ATMP界定接轨国际
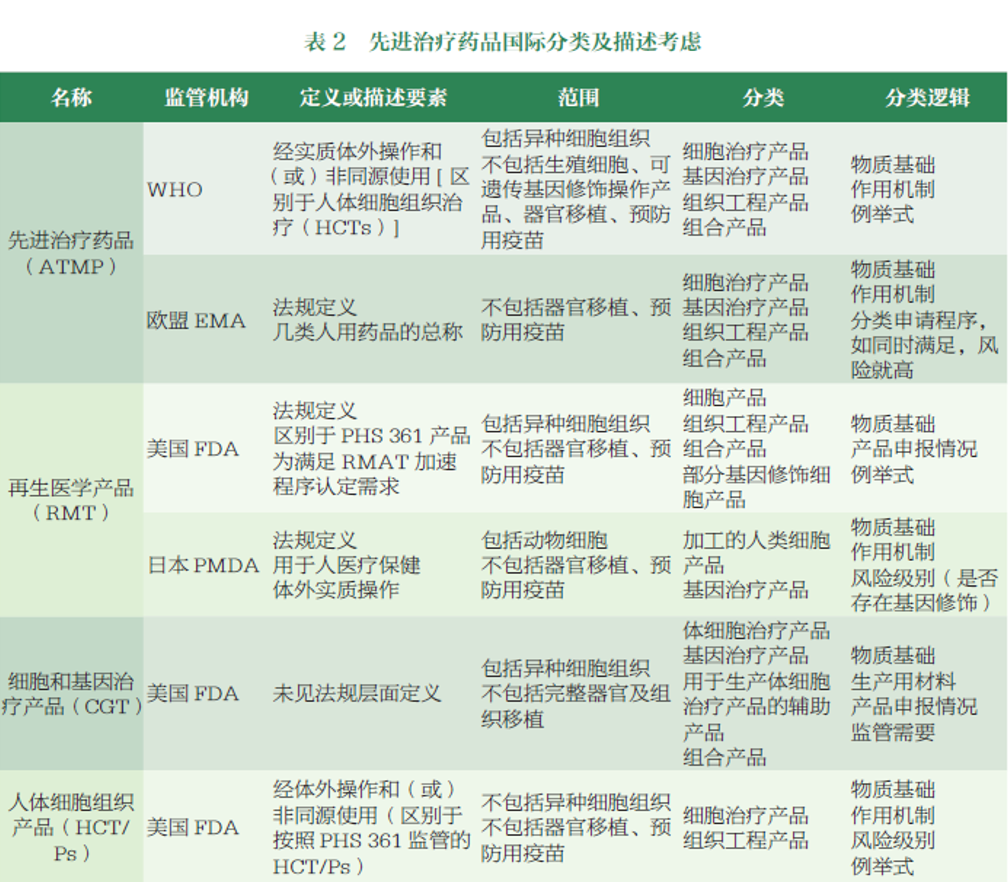
国家药监局自2019年启动《药品监管科学行动计划》以来,持续完善对先进治疗药品的全链条监管,但在该类药品的分类与定义方面,始终未能形成行业共识。2025年6月,《先进治疗药品界定与分类指导原则(征求意见稿)》(以下简称《征求意见稿》)的发布,是国家药监部门首次对ATMP作出清晰定义,明确将其分为细胞治疗药品(CTMPs)、基因治疗药品(GTMPs)及其他三大类。
研发客采访业内专家们表示,这一界定对规范监管、指导企业研发和接轨国际均具有重要意义。从监管层面看,结束了以往ATMP概念模糊、归类分散的局面,例如将细胞治疗药品细分为体外基因修饰、非基因修饰细胞药品两类;从企业层面而言,清晰的分类标准能够为药品研发立项、技术路线选择提供明确指引。

来源|CDE官网
更重要的是,此次界定搭建了我国ATMP监管与国际接轨的桥梁。
首先,名称上借鉴了WHO和EMA的先进治疗药品(advanced therapy medicinal products,ATMPs)表述,范围则整合参考了EMA、FDA等国际监管机构的分类框架。泰格医药高级副总裁、首席医学官、原CDE资深临床审评专家陈霞博士指出,这一接轨有利于我国研发与国际注册的对话衔接,打破国际多中心临床研究的跨区域监管壁垒。
来源|《我国先进治疗药品的范围及分类研究和建议》
不过,部分分类方式仍与国际存在差异。博济医药首席科学家、原CDE生物制品部审评专家万志红博士举例说明,以CAR-T疗法为例,其活性成分是经体外基因编辑修饰的细胞,兼具细胞产品与基因产品的双重属性,但《征求意见稿》明确依据发挥治疗作用的核心活性成分进行分类,将其界定为细胞治疗药品。这一分类与部分国际监管机构的分类不同,FDA、EMA、WHO等通常将基因修饰细胞纳入基因治疗产品的范畴。
值得注意的是,《征求意见稿》在常规细胞和基因疗法(CGT)的基础上拓展了范围,增设“其他”类别,将新型载体、细胞外囊泡等递送系统药品,以及组织工程药品等纳入其中,为各类创新性先进治疗技术预留了空间。
考虑到先进治疗药品技术迭代速度极快,陈霞建议在正式稿中设立定期修订机制,为体内基因编辑、合成生物学疗法等新兴技术模式预留归类空间,确保ATMP的界定能够动态适配行业发展。参考国际做法,EMA通过定期发布并持续更新ATMP科学分类推荐清单,收录产品分类界定实例供行业参考。
此外,优锐医药注册事务副总裁付洁鹰还建议,正式稿可以增设ATMP认定程序,企业可依此向药监部门提交申请,以获得对其产品属于先进治疗药品及具体分类的认定。他建议可以通过沟通交流方式或者类似申请附条件批准等方式推进这一程序。
ATMP加速路径不能操之过急
对企业而言,清晰的ATMP定义与范围界定是关键前提。产品一旦被认定为ATMP,企业更关心的是能否享受专属的加速审评路径。
参考国际经验,多个国家和地区已针对同类产品设立专属加速审评机制。美国设立再生医学先进疗法(RMAT)认定,申请产品首先需满足再生医学疗法定义,可以凭借较少早期疗效数据即可获得认定,相比突破性治疗所需的完整数据要求更低;且获得RMAT认定后,企业与FDA的早期沟通交流机会更多,能有效加速产品开发进程。此外,欧盟EMA设有RMAT分类程序,日本则推出SAKIGAKE认定等加速审评制度。
当下国内是否有必要为ATMP设立专门的加速路径?付洁鹰认为,我国的ATMP仍在逐步探索阶段,在ATMP分类和范围尚未完全明确的前提下,建议应结合实践情况逐步优化推进。
他认为,我国ATMP领域仍存在较明显的同质化问题,例如超过50%的CAR-T疗法临床试验聚焦在CD19靶点,因此,ATMP加速路径的审评标准不能仅以“是否为ATMP”为依据,还需结合具体临床数据、临床需求紧迫性等因素综合评估。
目前,我国药监机构已建立突破性治疗、优先审评审批、附条件批准等加速审评路径。陈霞认为,现有路径已能满足ATMP的审评需求,其实际加速效力并不逊色于国际上的RMAT机制。
万志红也给出实操建议:尽管目前暂未对ATMP设立特殊审批通道,但企业可充分利用CDE已发布的加快产品上市相关技术指导原则。她表示,目前我国已上市的10款细胞基因治疗产品,100%采用了至少一种加速上市途径。
值得关注的是,CDE于2025年9月发布《先进治疗药品(ATMPs)沟通交流中I类会议申请及管理工作细则(征求意见稿)》,进一步提升了ATMP沟通交流的效率。该细则明确了ATMP适用I类会议的三种情形,分别是纳入突破性疗法的ATMP、基因治疗类ATMP,以及进入关键临床阶段的ATMP。
陈霞特别强调了第三种情形的重要性,在完成探索性试验后、确证性试验启动前、确证性试验期间及上市申请前的沟通需求,都可以纳入I类会议范畴。“这相当于为所有进入关键研究阶段的ATMP开辟了绿色通道,是行业期待已久的‘加急沟通通道’。”她表示。
除了优化沟通机制,陈霞还建议尽早成立专门的ATMP审评部门。目前,我国先进治疗药品的审评工作主要由CDE下设的生物制品药学部、药理毒理学部、生物制品临床部等多部门联合开展。她指出,审评人员需同时兼顾常规品种与ATMP审评,易导致精力分散,进而可能影响审评的深度与专业度。
国际上,美国、欧盟均已单独设立ATMP审评机构。EMA成立了专门的先进治疗委员会(CAT);FDA CBER设立治疗性产品办公室(OTP)负责先进疗法审评,同时组建先进技术团队(CATT),通过CATT会议为企业与FDA提供非正式沟通渠道。
ATMP技术指南拼图渐成
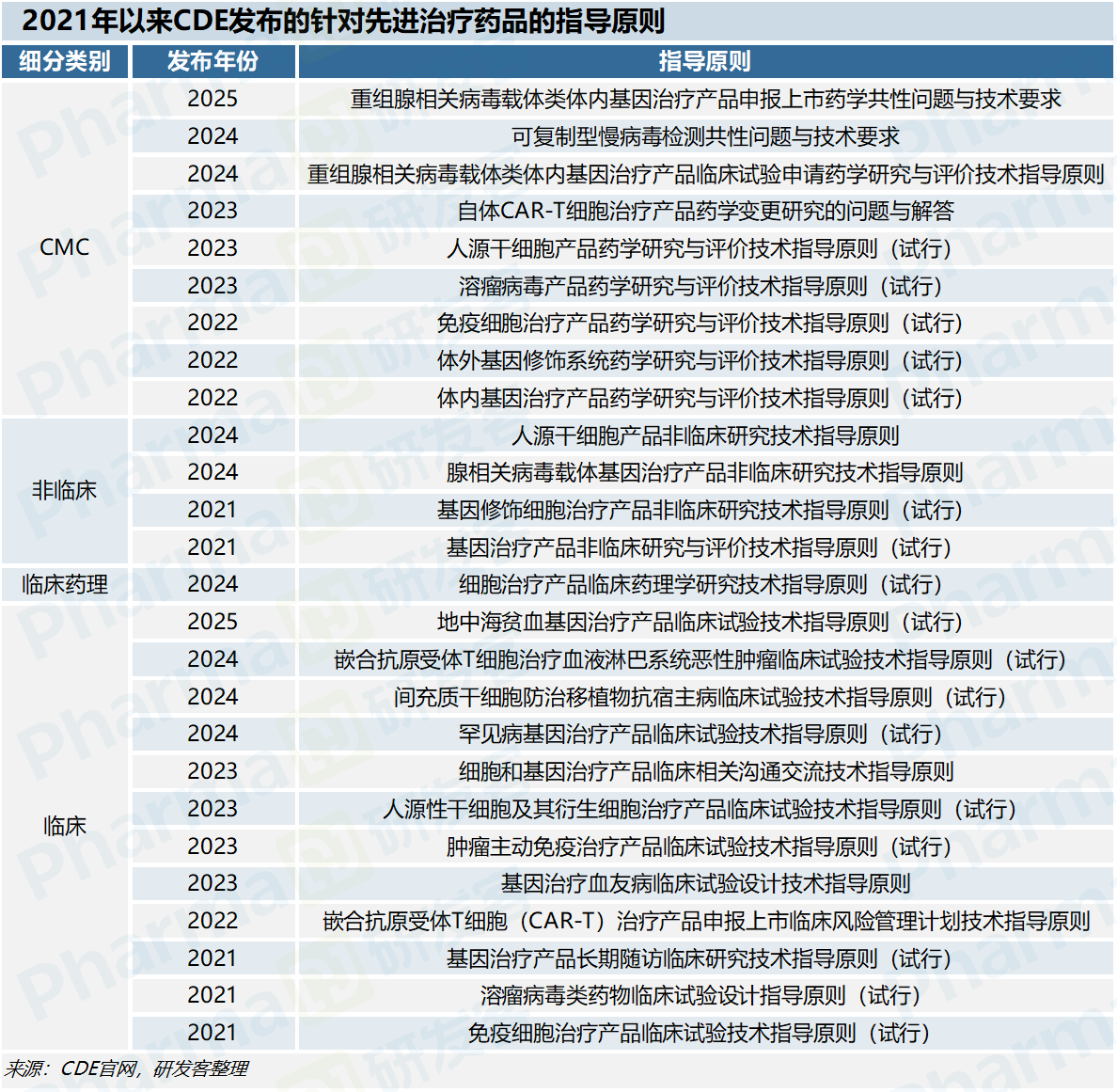
CDE自2021年起累计发布26项针对先进治疗药品的技术指南,覆盖了CMC、非临床、临床药理及临床等全链条关键环节。
例如腺相关病毒载体基因治疗产品的三项指南,分别从药学共性问题、药学研究、非临床研究三个维度,为这类高风险产品提供研发参考。陈霞认为这标志着我国先进治疗药品监管正从“事后评判”转向“过程指导”,不仅能帮助企业在研发早期规避关键技术风险,更能显著提高后续正式申报的通过率。
她进一步强调,2021年至2025年间,CDE通过高效的标准体系构建,将欧美监管机构10年完成的监管框架搭建历程缩短至5年,已形成覆盖“药学–非临床–临床–GMP–变更”的全生命周期监管技术矩阵。
展望下一步,陈霞建议可以将“技术指南+发补案例”整合为动态数据库,采用类似EMA Q&A的实时更新模式。此外,她还建议可以建立ATMP审评的专家共识集群,针对不同亚类产品发布更具体的技术审评要点。
付洁鹰强调,当前针对腺病毒载体、干细胞、CAR-T等主流先进治疗产品的技术指南已相对完善,未来可以向溶瘤病毒、体内基因编辑等新兴技术领域延伸,推动监管从被动审评走向主动指导。
关注IIT与注册临床的衔接
2025年发布的另一项ATMP相关的重磅政策同样值得关注,即《生物医学新技术临床研究和临床转化应用管理条例》(国令第818号)。
818号文确立的“备案+审批”双轨制,为干细胞、CAR-T、体内/体外基因编辑等尚未达到药品注册标准的早期ATMP,明确了从非临床研究、临床研究到转化应用的完整路径。通过该路径获批的技术由国务院卫生健康部门审核批准,其审批路径独立于NMPA的药品注册路径,且获批资质与《药品注册证书》具备同等法律效力。
不过,对于这些ATMP早期技术转化到药品注册的应用,即利用IIT(研究者发起的临床研究)数据支持药品注册审评,目前具体要求和流程尚未明晰。818号文只明确了国家卫健委会与药监局将尽快制定生物医学新技术与药品的界定指导原则。
万志红表示,药监部门已开展相关探索。2023年发布的《人源性干细胞及其衍生细胞治疗产品临床试验技术指导原则》中,专门提及“干细胞备案研究临床结果用于药品注册审评的评价要点”,明确了核心条件,包括IIT数据与IND申报数据的药学一致性和可比性,研究数据的可靠性和完整性,以及数据已显示初步临床获益优势。
陈霞建议,企业若计划利用IIT数据支持注册临床试验申报,在设计IIT之初就应前瞻性参照《药品临床试验质量管理规范》(GCP)及ATMP相关技术指导原则,确保研究数据(如细胞制备标准、疗效评价指标、随访计划)的质量和完整性,以满足未来注册申报的要求。同时还需及早与CDE沟通,明确哪些IIT数据计划用于注册申报,以及还需要补充哪些研究,实现IIT和注册临床试验的无缝转化。
参考资料:
卢加琪,刘丹,寇雅真,等.我国先进治疗药品的范围及分类研究和建议[J].中国食品药品监管,2024,(05):10-25.
编辑 | 姚嘉
yao.jia@PharmaDJ.com
访问研发客网站,深度报道和每日新闻抢鲜看