• FDA局长Marty Makary称,这是其推动的三大里程碑监管改革之一;
• 对临床终点大多为软终点的疾病领域的影响较大;
• 已有企业开始着手调整研发策略;
• Ⅱ期临床试验的战略地位将显著提升;
• 调整默认审批标准,并不意味着FDA不再要求药企开展两项临床试验。
今年2月,FDA局长Marty Makary在《新英格兰医学杂志》(NEJM)发文,宣告将“一项稳健且设计良好的关键性试验+确证性证据”确立为新药上市审批的默认标准(default standard)。他本人还向媒体表示,将药品上市默认要求的两项完整对照临床试验精简为一项,是其履职一年来推动的三大里程碑监管改革之一。
两项III期关键临床数据的要求,是FDA自上世纪60年代以来逐渐形成的审批惯例。而近年来,FDA早已在实践中采用“单项关键临床试验+确证性证据”的路径。
前FDA资深临床审评员、礼邦医药首席科学官肖申博士认为,明确这一默认规则将直接引导行业的行为走向。“以前企业可能会优先考虑两项临床试验,重申新标准后,企业会思考我们能否仅靠一项试验获批?整体证据链是否足以支撑仅凭一项试验上市?”
软终点领域影响更为突出
正如Marty Makary在NEJM文章中所提及的,近年来肿瘤和罕见病领域的多数药品均依托单项临床试验获批上市。
FDA曾针对2020~2022年批准的罕见病新药进行回顾分析(见下图),结果显示,其中80%(32/40)依靠单项试验+确证性证据获批。研究机构AgencyIQ的数据显示,2011至2019年,60%获批的抗肿瘤药物依据单项关键试验;2020至2023年间,约60%获批的新分子实体同样以单项关键试验为依据。
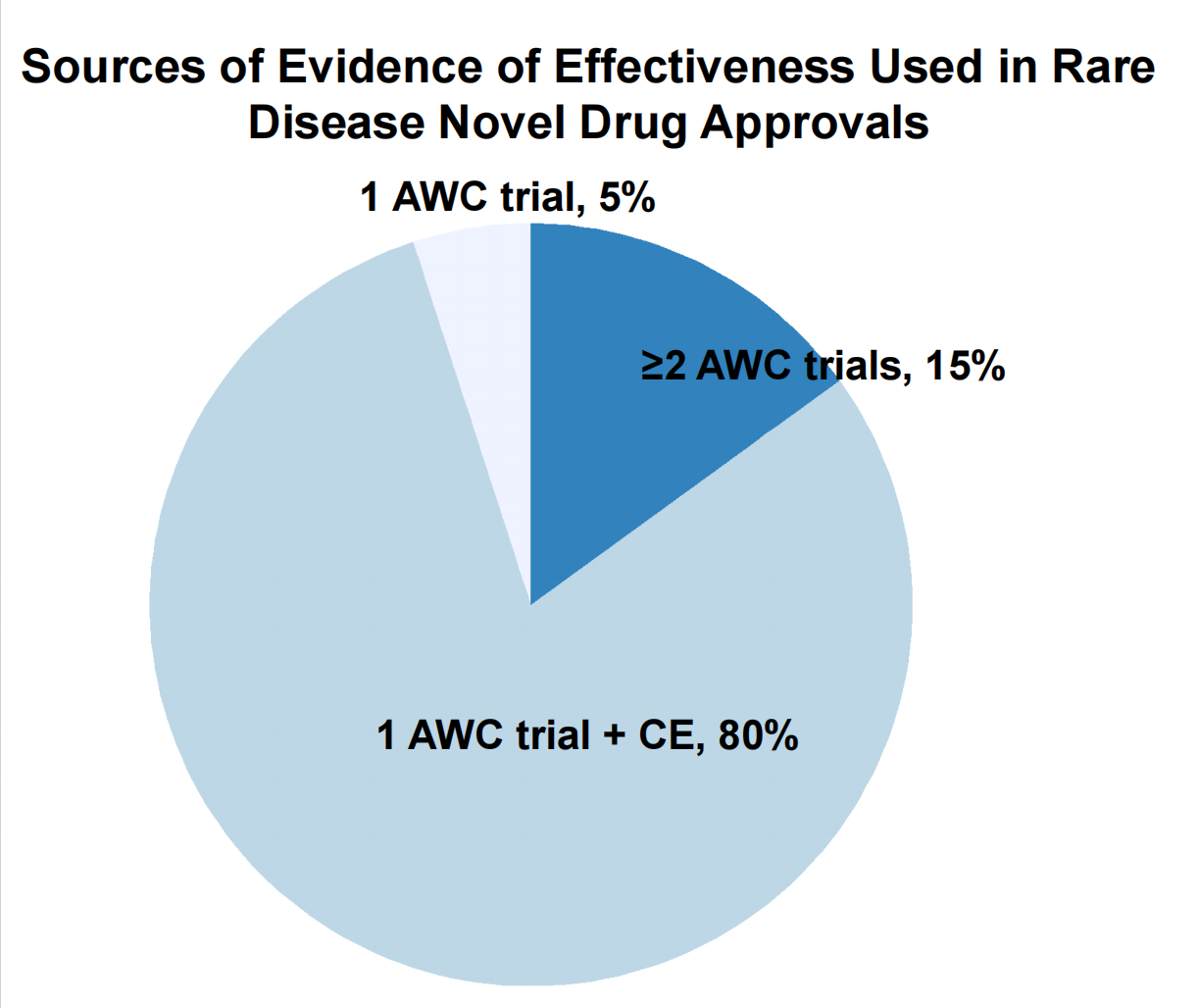
来源|FDA官网
这主要得益于近年来生物标志物技术日趋成熟、疾病机制研究更加清晰,临床试验统计方法也更为精进,肿瘤药物如EGFR、PD-1/L1等分子机制明确,OS、PFS等硬终点指标明确,单一试验提供的疗效证据更加稳定可靠,罕见病方面患者基数小、样本有限,特别是一些患者基数极小的超级罕见病,开展两项临床试验几乎不具备可行性。
因而,肖申认为,此次审批标准调整对肿瘤、罕见病药物的影响相对有限。他还举例,在他曾工作的FDA心脏病、肾病部门,如果临床主要终点是死亡率、心肌梗塞、中风等严重风险指标,也往往只做一个关键临床。

前FDA资深临床审评员、礼邦医药首席科学官
肖申博士
而真正受影响较大的将是眼科、神经、免疫等领域,他表示。这些疾病领域的新药上市申请,此前普遍被要求提供两项及以上关键确证性临床试验数据。例如,罗氏开发的治疗多发性硬化症的BTK抑制剂fenebrutinib,需待FENhance1和FENhance2两项关键研究数据全部公布后,才能递交上市申请;强生的卢美哌隆在2025年获批拓展适应症,用于重度抑郁症辅助治疗,仍须依据两项关键III期研究Study 501和Study 502。
肖申解释道,因为这些疾病领域的临床终点大多为软终点,容易受到患者自身状况和外部环境干扰,临床数据稳定性不足,进而进一步影响试验结果的判定。例如中枢神经系统(CNS)领域常用的认知功能评分量表、行为学评估等终点,以及眼科药物常用的最佳矫正视力、黄斑厚度、视野缺损等临床终点,均是临床试验中常见的软终点。
“针对这些稳定性不高的软终点,此前FDA往往会要求开展两项临床研究。而新的默认标准发布后,可能会有部分企业对这些疾病领域的新药研发重新调整策略。”肖申表示。
目前已有企业开始着手调整研发策略。Reunion公司在其官网公开表示(见下图),其治疗重度产后抑郁症(PPD)药物RE104在Ⅱ期临床试验中已取得顶线积极结果。该公司已经与FDA沟通并获得反馈,计划于2026年仅开展一项关键III期临床试验;若此项试验获得成功,则可凭借该关键临床数据支持RE104针对PPD适应症的上市申报。

确证性证据布局成关键
新默认标准确立之后,不止是临床研究策略,确证性证据(confirmatory evidence)的规划和设计也将成为企业重点考量的内容。
2023年9月,FDA发布指南“Demonstrating Substantial Evidence of Effectiveness Based on One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence”《通过一项设计充分且良好对照的临床研究和确证性证据证明有效性的实质性证据》,列出了上市申请中常用的确证性证据的类型:同类药物的临床数据、机制或药效学证据、同一药物在其他适应症的临床证据、真实世界证据(RWE)。
其中,机制或药效学证据是应用最广泛的类型,具体包含PK/PD、生物标志物和药物作用机制数据。
前述FDA所作2020~2022年批准罕见病新药回顾分析中,依靠单项试验+确证性证据获批的有32项,这其中有18项引用了机制或药效学证据:9项引用药效学生物标志物数据,另外9项引用药物作用机制数据。(见下图)
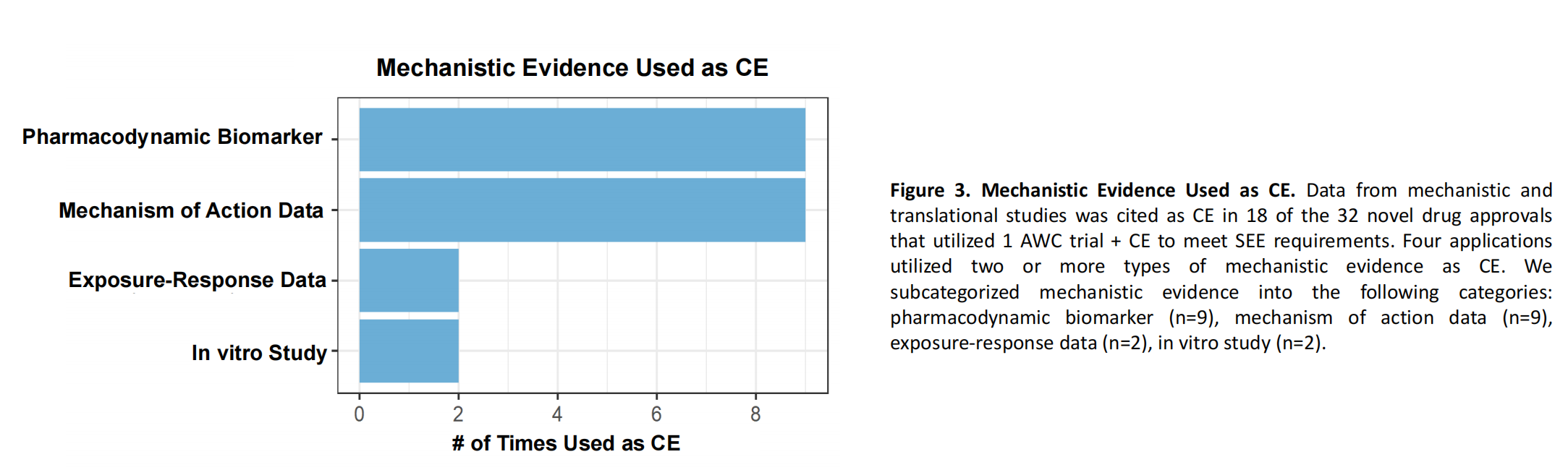
来源|FDA官网
由于机制或药效学证据多来自临床前、Ⅰ期及Ⅱ期临床阶段,肖申认为,Ⅱ期临床试验的战略地位将显著提升。
他强调,常规的Ⅱ期试验只是初步验证疗效,为Ⅲ期大规模研究提供依据,不具备独立确证疗效的效力;而如果将Ⅱ期数据作为确证性证据,与Ⅲ期数据联合用于上市申报,企业必须提早布局,在Ⅱ期阶段就采用更严谨、全面的试验设计。例如,更审慎选择临床终点,优先采用 FDA 指南认可、能真实反映临床获益的评价指标。
RWE是另一重要的确证性证据来源。NEJM文章也指出,单项关键试验审批改革,将与强化上市后数据收集(Postmarket Initiative)的政策同步推进。
不过需注意的是,RWE更多应用于已上市药物的适应症拓展、给药剂量优化等场景。例如,2019年,哌柏西利依据RWE拓展男性乳腺癌适应症,这也是首个仅采用RWE获批新适应症的药物。据 DIA期刊一项研究统计,2022~2024年FDA批准的218项获批药品标签拓展(以适应症拓展为主),有23%~28%比例获得了RWE支持。相对而言,目前全球范围内,以RWE直接作为新药首次上市的确证性证据获批的案例仍非常有限。
一个还是两个?并不绝对
回溯美国药品法规,1962年《Kefauver-Harris修正案》正式确立核心原则:新药上市须提供实质性证据(substantial evidence)证明疗效。在70年代出现了某些一项试验成功、另一项试验失败的情况,在抗抑郁药、抗心律失常药等领域尤为突出,FDA为避免偶然性,在80年代逐渐形成监管惯例,即企业需要提供两个III期关键临床结果、方可支持药品上市。
这一惯例从90年代开始转变。FDA在1998年发布《FDA现代化法案》配套指南“Providing Clinical Evidence of Effectiveness forHuman Drug and Biological Products”,提出一项充分且良好对照的临床试验,辅以确证性证据,即可作为证明药物有效性的审批依据。2019年,FDA 又对1998年版指南进行了进一步补充与扩展。
“FDA对新药审评的核心,从来不是试验数量的堆砌,而是依靠实证结果验证药物的真实临床价值。” 肖申表示。
他以P值举例,“过去在和企业讨论是做两项还是一项关键临床时,FDA就明确表明如果一项临床试验证明有临床意义的改变结合具备有说服力的P值(persuasive P value),可以不需要做第二个临床试验。所以将来,对常规的P<0.05在单一关键试验中是否满足获批要求,FDA可能会要求更严格的阈值,如P<0.01,甚至P<0.001,对不同的疾病也许会有不同的要求。”
NEJM文章中也重点强调,调整默认审批标准,并不意味着FDA不再要求药企开展两项临床试验。若药物作用机制模糊或缺乏特异性,试验终点指标不稳定或使用替代终点,抑或试验存在设计缺陷等情况,FDA仍会要求企业补充开展充分且良好对照的临床试验,甚至要求开展三项及以上试验。
文章中还特别指出,仅开展一项临床试验后,FDA对关键临床试验的审评要求会更高,会严格审评试验设计的每个环节,重点关注对照组设计、临床终点、效应量以及统计学研究方案。
编辑 | 姚嘉
yao.jia@PharmaDJ.com
访问研发客网站,深度报道和每日新闻抢鲜看