• 通过提升AI监管门槛,倒逼医药产业进行数字化转型,淘汰落后产能,推动中国从制药大国向制药强国迈进。
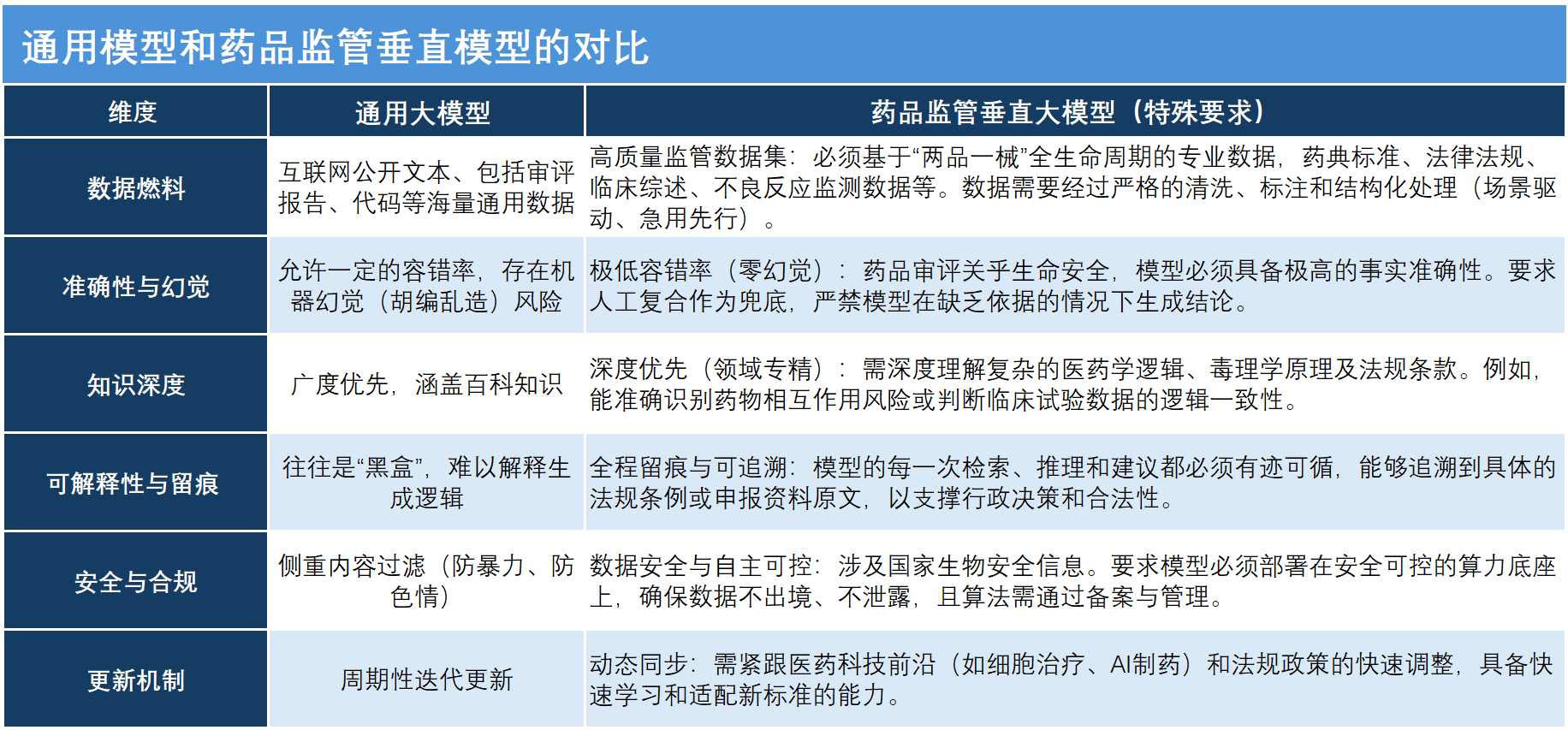
• AI不再是聊天机器人,而是具备了药品监管专业知识的专用工具。与通用语言类大模型相比,药品监管垂直大模型的区别在于底层数据库来源,整体具有高度封闭性和极强的专业性。
• 药品审评审批关乎生命安全,模型必须具备极高的事实准确性。要求人工复核作为兜底,严禁模型在缺乏依据和训练下生成结论。
• 药品最终批准权毫无疑问必须始终由人类审评员掌握。这一机制并非用AI取代人类,而是通过人机协同,实现1+1>2的效能提升。
• 相比美国FDA的“敏捷试点”和欧洲EMA的“伦理审慎”,中国正在探索走一条顶层设计先行、基础设施集约化、监管与产业协同的独特道路。
4月2日,国家药监局(NMPA)正式发布《关于“人工智能+药品监管”的实施意见》(以下简称《实施意见》),标志着我国药品监管正式告别电子化时代,全面开启以数据要素为驱动、以人工智能为核心的“数智化”新征程。
这一政策背后有着怎样的战略考量,NMPA在国际药品监管AI应用中处于什么位置?带着这些问题,研发客专访了中国药品监督管理研究会会长张伟,以及中科计算技术西部研究院研究员、图灵·达尔文实验主任赵宇教授。
战略考量:从信息化向数智化跨越
张伟告诉研发客,这一政策并非孤立的技术升级,而是国家在医药监管领域的一次战略性重塑。换言之,利用人工智能技术重构药品监管的底层逻辑,实现监管能力的现代化。

中国药品监督管理研究会会长 张伟
如何重构?第一是推动监管模式从事后被动转向事前主动。“传统的监管往往依赖于事后抽检或不良事件报告。《实施意见》明确提出要利用AI构建风险监控智能体,通过对生产、流通、使用全链条数据的实时分析,实现风险的动态监测与主动预警。”
张伟以疫苗举例。疫苗生产过程的物联感知数据,可以实时分析、实时预警,战略目标是将风险隐患消除在萌芽状态,构建“数智驱动、智能敏捷”的治理新格局。
第二是构建“人机协同”的新型生产力。面对海量的审评审批任务和复杂的创新药械,单纯依靠人力已难以满足需求。《实施意见》提出构建人机协同智能审评审批体系,利用大模型辅助资料审查、报告生成和知识检索,张伟认为其战略目标即“在守住安全底线的前提下,大幅提升审评审批效率,解决行业长期关注的药品上市慢痛点”。
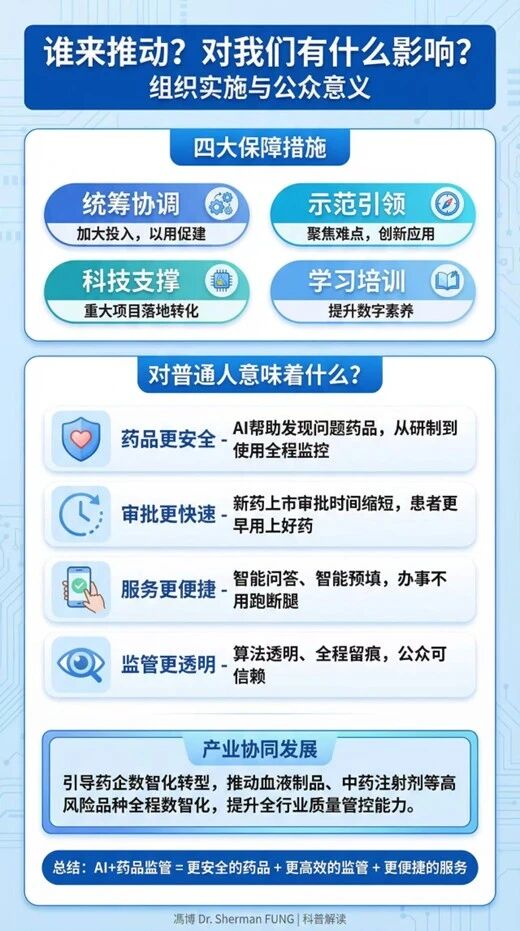
第三是以监管标准,倒逼产业升级。张伟特别提到,《实施意见》中强调的“促进监管与产业数智化协同发展,特别是针对血液制品、中药注射剂等高风险品种,要求引导企业提升全过程质量管控能力”,“战略目标就是通过提升监管的数智化门槛,倒逼医药产业进行数字化转型,淘汰落后产能,推动中国从‘制药大国’向‘制药强国’迈进”。
《实施意见》发布既是落实国家“人工智能+”行动的必然要求,也是对国家战略部署的具体跟进,旨在抢抓人工智能发展带来的重大战略机遇,与国务院此前印发的《关于深入实施“人工智能+”行动的意见》形成了呼应。
同时,发布的这一时间节点也立足于“十四五”智慧监管成果的深化与迭代升级。2026 年是“十五五”规划开局之年,《实施意见》既可以系统总结过去五年药品监管信息化建设经验,也能利用大模型等新技术,为未来5至10年的监管现代化绘制蓝图。
而从产业的角度来看,《实施意见》的推出是技术爆发与产业需求的双向奔赴。张伟认为,近年来医药产业创新活跃,新业态层出不穷,传统监管手段面临挑战,迫切需要新的监管工具,近年来生成式人工智能、大数据等技术快速迭代,已使得在监管中大规模应用AI具备可行性。
两步走战略:从筑基期到升华期
《实施意见》明确了清晰的两步走战略目标。张伟告诉研发客,这两个阶段的目标并非简单的线性时间推移,而是体现了从技术应用与机制建立到生态重塑与格局形成的递进。“简单来说,到2030年的阶段是打地基、建框架、跑通流程,而到2035年的阶段是全装修、强内核、形成生态。”
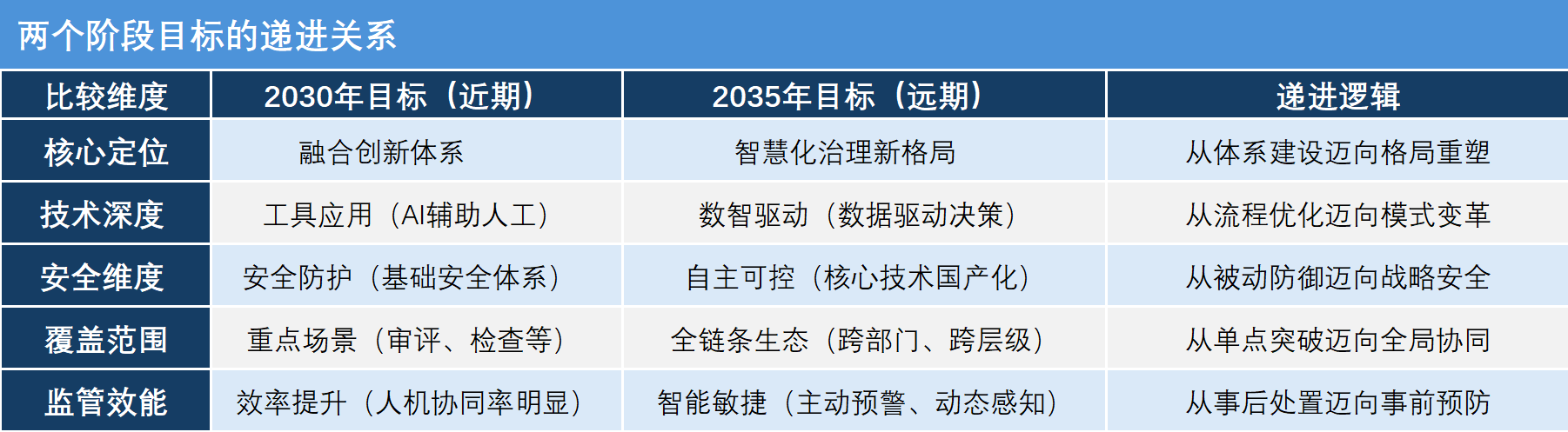
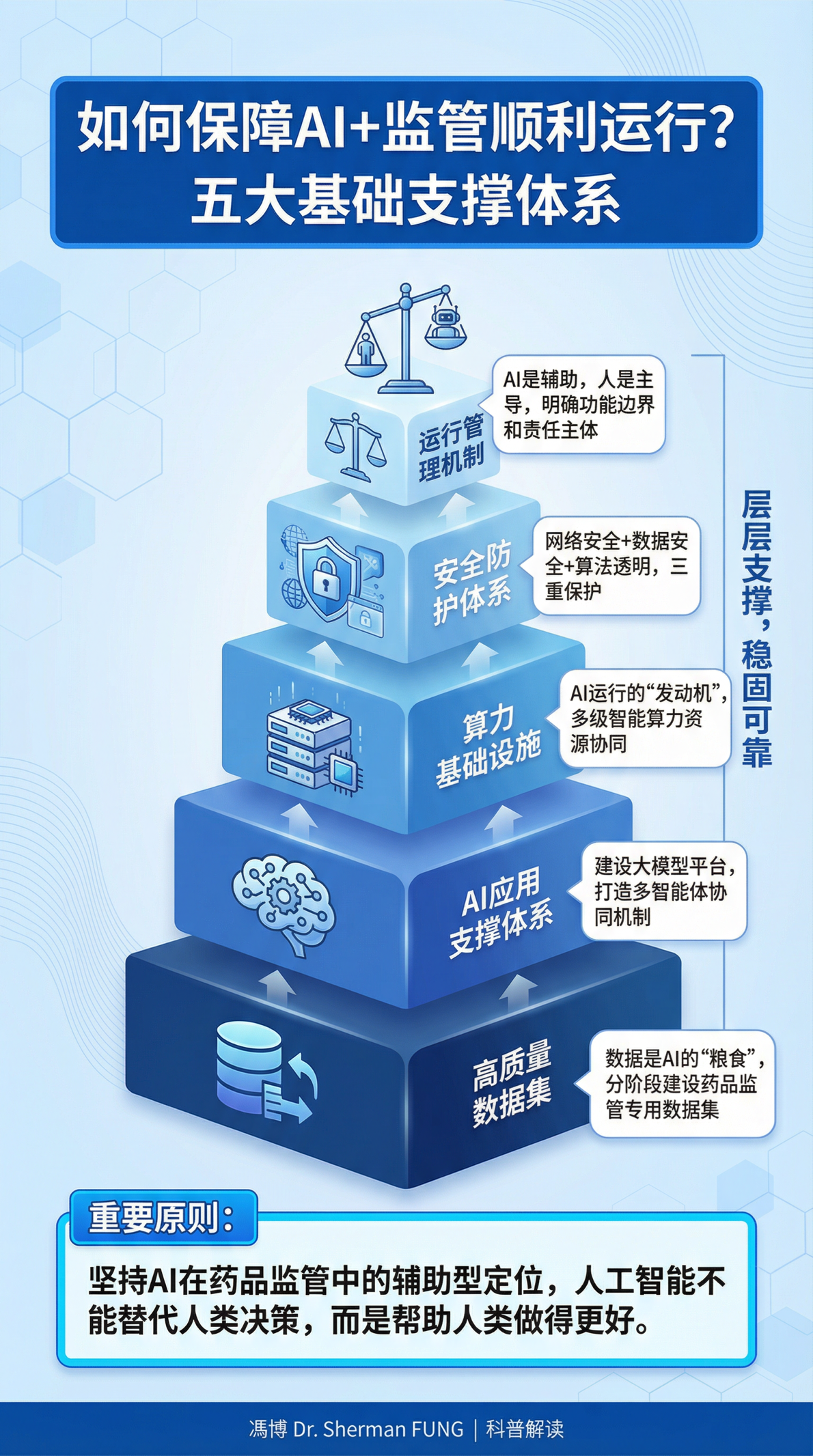
到2030年,初步构建融合创新体系,应用落地、机制形成、效率提升。具体包括:算力支撑底座更加集约高效,形成满足监管智能化需要的高质量数据集;形成垂直大模型和智能体,AI不再是通用的聊天机器人,而是具备了药品监管专业知识的专用工具;AI在审评审批、监督检查等核心场景中实现有效应用;确立“数智赋能、人工复核、全程留痕”的运行管理机制。
到2035年,基本形成智慧化治理新格局。具体包括:由2030年的辅助应用升级为数智驱动,数据和算法将成为决策的核心依据;特别强调自主可控,确保核心算法、数据和系统不受制于人;强调“生态协同”,形成政府、企业、社会多方联动的治理体系。
从记录员到总设计师
谈到《实施意见》里“以信息化引领药品监管现代化”这一核心指导思想,赵宇十分赞同。
他认为“引领”用得非常准确:“在第四次工业革命浪潮下,信息技术正在对原有政策解读、复杂监管场景认知、监管效率提升以及人力成本节省这四个方面,发挥着转折性、关键性的作用。” 人工智能作为信息化技术的重要载体,其核心价值就体现在助力监管效率提升和认知边界拓展上,为药品监管现代化提供强有力的技术支撑。

中科计算技术西部研究院研究员、图灵·达尔文实验主任 赵宇教授
“这意味着信息化不再仅仅是监管工作的记录员,而是成为了监管现代化进程的总设计师和发动机。”张伟说。
借助信息化,监管逻辑从事后灭火转向事前预警。通过大数据和AI模型对全生命周期数据进行实时分析,实现事前主动预警;生产关系从人力受限转向人机协同治理体系。确立“数智赋能、人工复核、全程留痕”的人机协同机制,重新定义了监管人员的角色,将他们从繁琐的重复劳动中解放出来,专注于复杂的决策与判断;产业升级以监管标准倒逼产业数智化。通过设定高标准的数智化监管要求,倒逼并引导医药企业进行数字化转型。
人机协同:AI辅助,人类决策
国家药监局已明确将“协同共建‘两品一械’审评审批大模型与智能体”列为首要重点任务,计划到2030年形成满足监管智能化需要的高质量数据集、垂直大模型和智能体。《实施意见》发布之时,该大模型的研发刚刚进入全面部署与加速建设期。
张伟特别强调:“‘两品一械’大模型的研发目前正处于筑基阶段,其核心难点不在于算法本身的先进性,而在于构建高质量的行业专用数据集以及建立人机协同的信任机制。”
“与大家较为熟悉的通用语言类大模型相比,药品监管垂直大模型的核心区别在于底层数据库的来源。通用大模型多基于互联网公开信息、各类随手可得的文本信息训练而成,而药品监管垂直大模型必须依托专属的垂类数据库,这些数据库除了政策明确的品种档案、企业信用档案、法律法规库、典型案例库之外,还涵盖过往的监管信息、审评数据等,整体具有高度封闭性和极强的专业性。”赵宇教授说。
“基于这样的专属数据库训练而成的大模型,还需要具备针对性的算法能力,才能构建形成真正赋能审评业务的AI工具,大幅减少幻觉现象,输出的意见更具专业权威性,这也是药品监管垂直大模型最核心的特殊要求。”
关于人机协同,张伟强调:“智能审评审批体系的核心原则是AI辅助、人类决策,最终的批准权毫无疑问,并且必须始终由人类审评员掌握。这一机制并非用AI取代人类,而是通过人机协同,实现1+1>2的效能提升。”
在这一体系中,AI是强大的副驾驶,负责处理数据和提供导航;而人类审评员是手握驾驶杆的机长,负责最终决策并对公众健康的安全负责。
从具体分工而言,人工智能的角色是高效的初级分析师。AI负责处理海量信息、执行标准化任务和提供决策支持,具体包括信息处理与摘要、形式审查与校对、风险预测与提示、知识检索与问答。
“它将审评员从繁琐、重复的劳动中解放出来。”张伟强调:“人类审评员的核心价值在于AI无法复制的专业智慧、经验直觉和伦理判断。最终是否批准一个药品上市,必须由具备法定资格的审评员在综合评估所有信息后做出判断并承担责任。同时,审评员还需要复核与监督AI的输出,处理例外与伦理问题。”
赵宇也认同“整个机制的核心仍然是以人为主导,最终的决策权限始终掌握在人类手中。”正如医院的诊疗工作,机器学习可以给出专业建议,但医生的处方权始终属于人类医生。
“人工智能的优势体现在文本处理能力,以及在审评工作的深度和广度上,远远超出人类的能力范围。正如CDE此前提出的,我们既可以‘向AI要人’,借助人工智能提升审评效率、节省人力成本,也可以‘向AI要科学’,通过人工智能拓展我们对审评工作的认知宽度和深度,助力我们在监管决策中达到更高水平。”
中国AI审评从跟随者变成并跑者
谈到与国际同行的比较,张伟认为,中国在AI应用的版图中正从跟随者向并跑者、甚至在部分领域向领跑者转变。相比美国FDA的“敏捷试点”和欧洲EMA的“伦理审慎”,中国正在探索走一条顶层设计先行、基础设施集约化、监管与产业协同的独特道路。
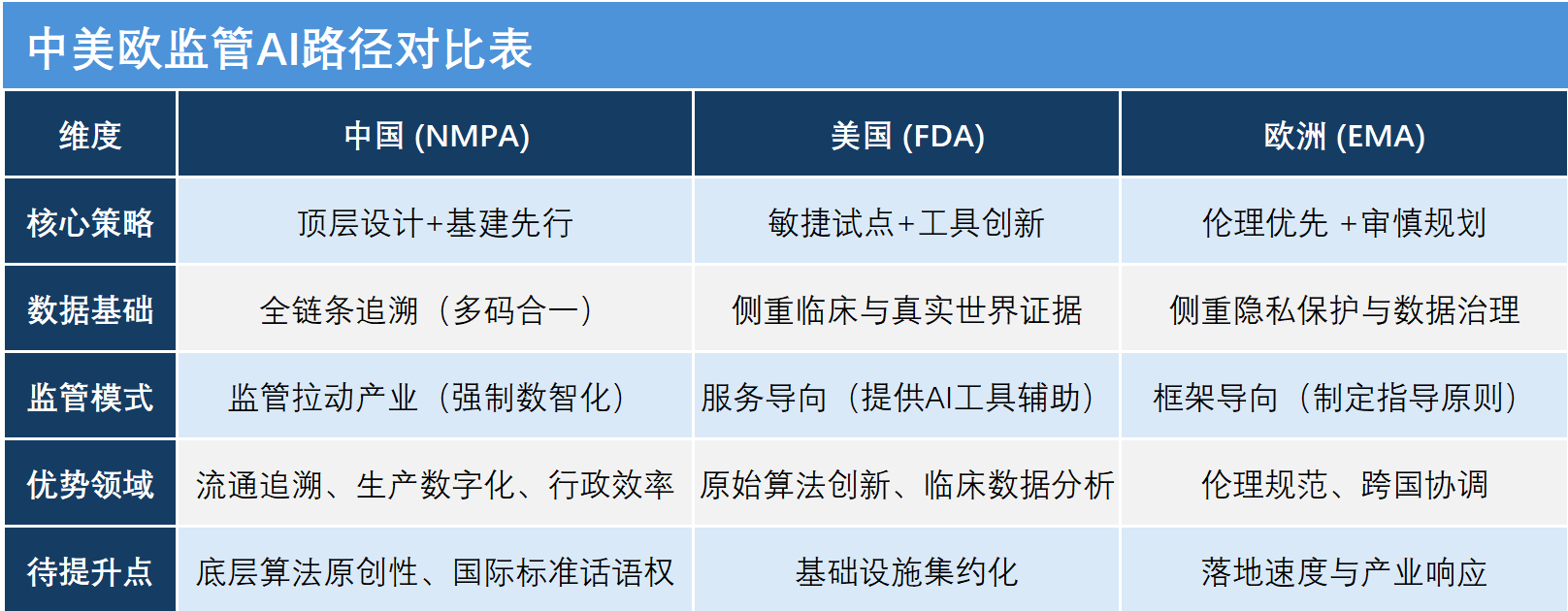
同时,张伟也坦承中国需要加快追赶的领域:“首先是原始创新与底层算法,目前我们在应用层做得很好,但在底层算法模型上与欧美顶尖水平仍有差距。其次是国际协调与标准话语权,中国在AI监管的国际标准制定中的话语权仍需加强。第三是算法的可解释性与信任机制,如何让人类专家信任并理解AI的建议是一个全球性挑战。”
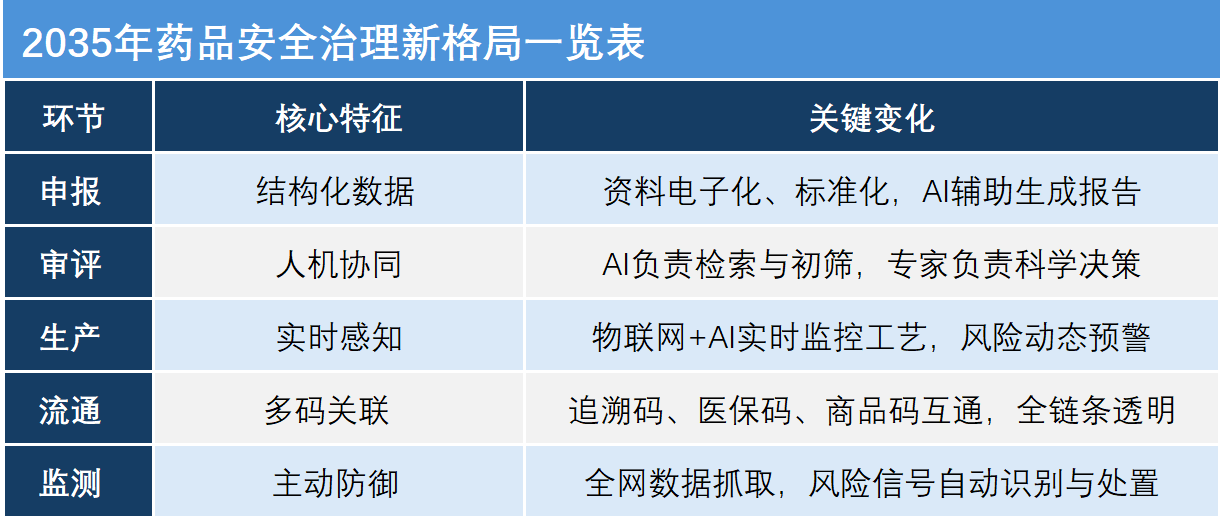
展望2035年,张伟描绘了一个普通药品从申报到上市的全流程将经历的深刻“智”变:
在申报与审评环节,药品注册申报将不再是堆积如山的纸质资料,而是标准化、结构化的数据流。企业提交的数据将直接符合“机器可读”的标准,AI智能体会在申报端即时进行预审查,审评审批大模型会立即进行智能分类并自动分配任务。
在研制与临床试验环节,监管将深度嵌入到研发的全生命周期中。临床试验数据将实现电子化记录的规范化,监管部门的AI系统可以实时接入临床试验数据,自动监测数据的异常波动或潜在的造假行为。
在生产与制造环节,将实现非现场监管与现场检查的无缝融合。药企的生产线将与监管平台实时联网,AI通过物联网传感器、视频监控和图像识别技术实时分析关键工艺参数,一旦发现偏差立即预警。
在流通与使用环节,“多码合一”将彻底解决药品来源可查、去向可追的问题。每一盒药都将拥有唯一的“身份证”,AI会对每一批次的药品绘制动态风险画像,普通患者通过手机扫描药盒上的码即可看到药品的“前世今生”。
在上市后监测环节,将进入全域实时监测时代。AI将全天候扫描全网的投诉举报、社交媒体舆情以及医疗机构的不良反应报告,从海量的非结构化文本中敏锐捕捉潜在的风险信号,一旦确认风险,系统能基于追溯体系在几秒钟内精准定位受影响药品的具体流向。
赵宇也积极看待AI监管的未来:“药品的发现将不再依赖天才般的偶然灵感,而是会走向工程化的必然,就是通过数智技术的赋能,我们能够更精准、高效地推进药物研发。”
他预言:“2035年左右,研发一款新药也许只需要6~8年的时间,在全球范围内可能仅需1亿美元。这种高效研发模式正是智慧化药品安全治理新格局对药品从申报到上市全流程最核心的改变。若能成为现实,必然需要一套与高速研发同频共振的数字化证据运行机制作为支撑。
“随着‘数字理解生命’ 范式走向成熟,研发中应用的 AI驱动的生物学假设、虚拟患者画像,将不再局限于企业内部研发使用的数字化洞见,而逐步被监管认可为可评价、可量化、可追溯的正式数字化证据。”
他最后指出,这一转变有望分三阶段推进:完善现有证据体系、部分替代真实试验,最终在特定场景实现对传统试验的替代。


图解《关于“人工智能+药品监管”的实施意见》 制作|冯博士(Sherman)
我们还将详细拆解美国FDA与欧洲EMA联合发布的“Good AI Practice”(GAIP)十项原则,敬请关注。
编辑 | 姚嘉
yao.jia@PharmaDJ.com
访问研发客网站,深度报道和每日新闻抢鲜看