• FDA联合EMA发布的Good AI Practice(GAIP)十项原则,可视为“AI时代的质量管理规范”;
• FDA监管两大关键:AI用在哪儿,数据从哪儿来;监管的本质不是算力,而是责任。而责任,只能由“人”来承担。
• 影像学临床终点评估、患者筛选与分层等AI应用场景的监管要求非常高;
• AI只是助手,最终决策必须由人来做出,因为监管的本质不是算力,而是责任;
• 对于中国生物医药企业,前FDA资深审评员肖申博士提出三点应对AI监管的建议。
在制药行业这一强监管领域,历来有GLP、GCP、GMP等作为指引。针对AI制药(AIDD),美国FDA与欧盟EMA联合推出了专门为AI应用制定的实践指南(Practice)。
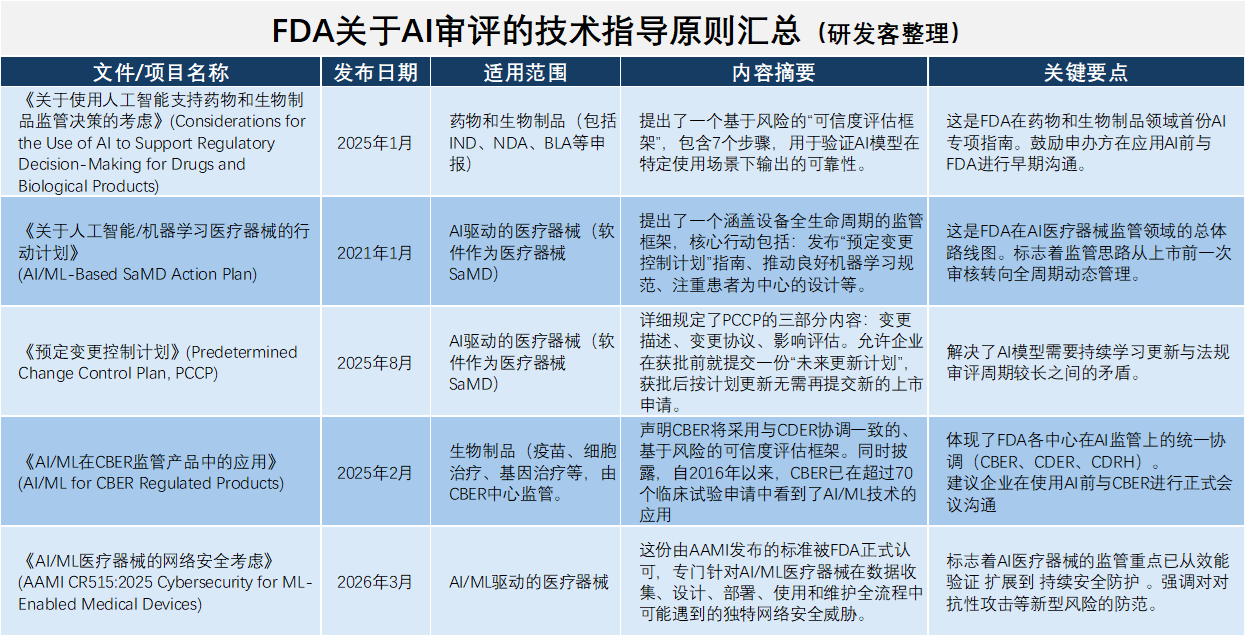
2025年1月,FDA发布了关于AI在药物开发中应用的指南(下称“AI指南”),2026年1月又联合EMA发布了“Good AI Practice”(GAIP)十项原则(见下图。FDA关于AI审评的技术指导原则汇总见文末)。这意味着监管机构正式认可AI在药物开发中的应用,并通过制定相应的规范,引导AI辅助的药物临床试验走向规范化和标准化。

来源|FDA官网
“GAIP传递了FDA的明确信号:鼓励企业在临床开发中应用AI,并将在审评环节采纳AI。”前FDA资深审评员、礼邦医药首席科学官肖申博士说,AI在药物发现前期的作用越来越大。但监管的态度很清楚,“前面怎么探索都可以,一旦要上人了,就必须严格评估风险。所以现在不是‘要不要用AI’的问题,而是‘在哪个环节用、怎么用、用什么标准来验证’的问题。”
指南两大核心:应用场景和数据来源
肖申将AI指南归纳为两个核心。
第一是应用场景(Context of Use,COU)。“监管不是一刀切。”肖申说,在早期研发阶段,如靶点发现、分子筛选,监管比较放松,因为风险低。但一旦进入非临床安全评估、临床试验、生产质量控制,监管要求就很严格。FDA的指南里反复强调:企业在什么场景下用AI,决定了企业需要接受多严的验证。
第二是数据来源。研究的数据是否可靠、有没有追溯性、是不是有代表性、能不能避免数据偏移,FDA对这些问题的审查非常严格。“监管现在就抓住这两点:AI用在哪儿,数据从哪儿来。这两条只要能把控住,AI就能真正帮到药物开发。”
GAIP十项原则
GAIP十项原则,“可以理解为AI时代的GCP,它告诉大家:申请人按照这个路径走,FDA就可以认可你的结果”,肖申表示。
第一是以人为本的设计。不能为了AI而AI,不能为了技术而炫技。一切要服务于患者利益和伦理,这个底线不能破。
第二是基于风险的方法。高风险场景,比如临床终点评估、剂量选择、安全信号监控,需要高强度的验证;低风险场景,比如文献整理、医学翻译写作,可以灵活一些。
第三是遵循标准。在AI开发与使用过程中,要遵守适用法律、伦理、科学、技术、网络安全和GxP等标准。
第四是明确使用场景。清晰界定AI的目标、适用场景、角色与范围,确保其在特定背景下提供可靠支持。
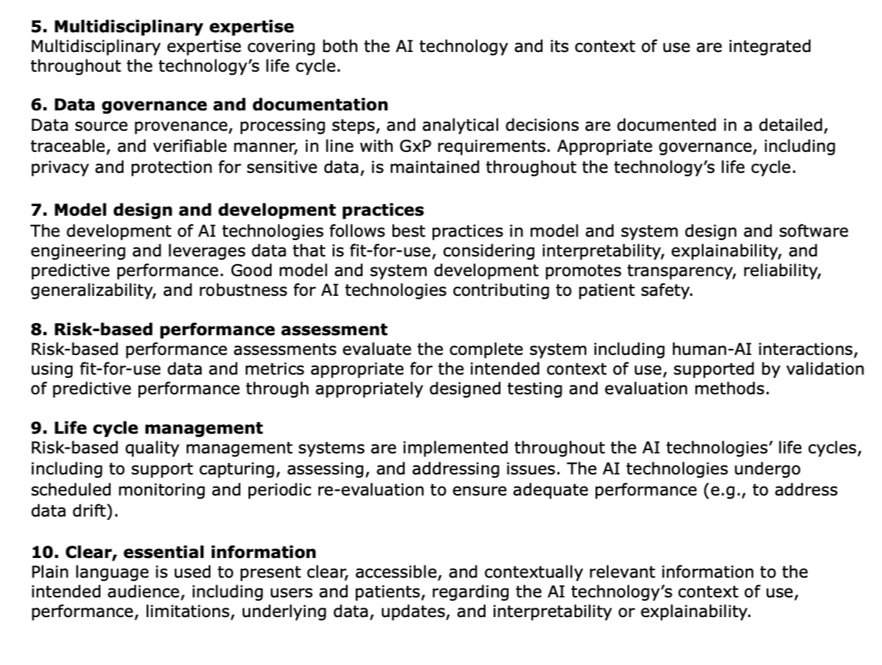
第五是多学科专业参与。AI不是IT部门的事。在AI系统开发、评估和实施过程中,需要有临床、统计、数据科学、软件工程、质量与药物警戒等多学科专家参与。
第六是数据治理与文档管理。建立稳健的数据治理机制,对数据来源、质量控制、访问权限和处理过程进行记录与跟踪,确保文档完整、可追溯。
第七是模型设计与开发实践。遵循良好的软件工程和机器学习实践,包括版本控制、可重复性测试、安全性与变更管理等。
第八是基于风险的性能评估。针对具体使用场景开展性能评估,包括偏差检测、模型漂移压力测试和验收标准评估,确保模型能够在预定情境中可靠运行。
第九是生命周期管理。AI模型不是一次性的。数据会偏移,环境会变化,模型需要持续监控、定期重测。在整个生命周期内持续监控AI系统的表现,控制风险,在重大变更后重新验证,并在适当时负责地退役模型。FDA需要对患者负责。
第十是清晰、必要的信息。企业不能给监管一个“黑盒子”。要向相关利益方提供透明、易懂的关键信息和解释,减少“黑箱”效应。“你越坦白,给的证据和论据越多,监管越放心。”(具体见下图,上下滑动查看)

FDA七步风险评估流程
结合AI指南,肖申把FDA评估AI应用总结出七个步骤,认为“从风险评估、模型设计、训练验证,到实施计划、监管沟通、文档记录,每一步都写得很清楚”。
“第一步至第三步属于初步评估与准备阶段”。第一步是定义使用场景(COU),明确AI在药物开发中的具体用途,例如患者筛选、影像终点评估或安全信号检测。第二步是评估模型对患者和决策的影响,分析模型输出对患者安全、疗效判断或监管决策的潜在影响,从而确定风险等级。第三步是确定模型的输入与输出,明确模型所需的数据类型、来源、处理流程,以及输出结果的格式和解释方式。
第四步是模型开发与训练过程的关键考量。这一步骤涉及多个技术细节,FDA特别关注模型的训练过程。
第五步是实施该计划。在模型开发完成后,申办方需在实际应用场景中实施AI模型,并确保其符合监管要求。
第六步是记录计划并评估执行偏差。申办方需记录AI模型开发与验证的全过程,包括模型输出的可信性评估结果,并讨论实际执行过程中是否出现偏差。
第七步是确认模型是否适用于该使用场景。最终,FDA要求申办方确认AI模型是否足够稳定、可靠,能够在其定义的使用场景下持续发挥作用。申办方需提交完整的生命周期管理计划,包括性能指标描述、基于风险的监控频率设定、模型重测触发机制等,并提前与监管部门沟通变更管理计划。
“企业不需要让监管完全理解你的算法细节,但你需要提供足够的证据,让监管相信用AI做的结果,在安全性、有效性、质量可控性上不亚于人工。你可以做人机对照,证明一致性;你可以做压力测试,验证鲁棒性。关键是,你得有证据。”肖申说。
哪些场景监管最严?
具体来说,有几个应用场景的监管要求非常高。
影像学临床终点评估方面,AI如果用来替代人工读片、判断疗效终点,需要严格的外部验证,要与人工读片对照。之前就有项目因为没提供充分的外部验证,被要求补做人工读片对照研究。
患者筛选与分层方面,AI用来决定谁入组、谁出组,会影响整个临床试验的人群构成。必须评估选择偏倚风险,证明不会系统性排除某些亚群。
安全信号检测方面,AI辅助发现不良事件信号,目前更多是辅助决策,不是自动决策。要关注假阳性、假阴性风险,要与传统规则系统做对比。
真实世界证据生成方面,用AI分析真实世界数据来支持适应症扩展,关键在于数据质量和混杂控制。模型透明度要求高,需要与随机试验结果形成一致性证据链。曾有项目因模型混杂控制不足,导致审评进度延迟。
CMC质量放行方面,AI用于生产过程中的质量检测,比如视觉缺陷识别,这直接影响批次放行决策。有项目因为模型漂移没及时发现,批次间差异扩大,被要求暂停自动放行。
肖申认为,这些场景的共同点是高风险、直接影响患者安全或疗效判断,因此,“验证必须覆盖真实使用环境,外部验证几乎不可省略,模型更新需受控,系统级风险评估比模型指标更重要。监管不是卡你,是帮你把风险控制住”。
数据偏移和幻觉问题怎么解决?
数据偏移分三种:特征偏移(输入特征分布变化,但标签规则不变)、标签偏移(输出标签的分布变化,但特征与标签的关系不变)、概念偏移(特征和标签的映射关系本身发生变化)。简单说,就是训练时的数据和实际用的数据不一样了,模型性能就会下降。监管要求企业建立持续监控和定期重测机制。“你不能一个模型用到底,要定期验证,发现偏移就要重新训练或调整。”
“幻觉”即AI编造不存在的东西,目前还没有完美解决方案,所以监管的态度是:AI辅助人,而不是取代人。
“特别是在高风险决策上,最终判断必须由人来做出。你可以让AI先筛一遍、算一遍,但关键结论要有人复核。人机结合,是目前最务实、也最被监管接受的模式。”肖申表示。
AI会取代审评员吗
FDA已经在做一些探索,把过去几十年的审评资料电子化、格式化,用来训练AI,帮助审评员做初步筛查、文献整理、常见问题判断。
“这就像年轻医生有了一个‘老专家知识库’,效率会提高。”不过肖申认为,“真正复杂的审评,比如权衡一个全新靶点的风险获益、判断一个临床试验设计的合理性,需要的是底层的医学知识、药理毒理功底、以及主动的逻辑思维。这些东西,目前的AI还做不到。”
肖申认为,可以把AI理解为一个非常聪明的助手,它帮你整理信息、提示风险、建议方向,但最终决策必须由人来做出。因为监管的本质不是算力,而是责任,“责任,只能由人来承担”。
基于以上分析,肖申对中国生物医药企业提出三点建议:
首先要早沟通,不要等做完了再去找监管。企业在设计模型的时候,就可以跟监管开会,确认验证方案。越早沟通,后面越顺。
其次,要建立多学科团队。AI不只是IT部门的事,还需要临床、统计、药理、毒理、质量、法规部门一起参与。“FDA的GAIP原则里专门有一条‘多学科专业参与’,这不是随便说说的。”肖申说。
第三,用监管能听懂的语言,讲清楚数据来源、验证结果、风险控制措施,不需要炫技,不需要把模型设计得特别复杂。
总结来看,AI模型在药物审批中的支持作用需要重点关注四个方面:数据质量、模型验证、可解释性、持续监测。未来,AI在药物研发中的应用将更加广泛,但也将面临更严格的监管。“对于企业来说,理解监管的逻辑、主动拥抱标准、坚持人本和透明,才能在AI制药这条路上走得更远、更稳。”肖申说。
编辑 | 姚嘉
yao.jia@PharmaDJ.com
访问研发客网站,深度报道和每日新闻抢鲜看