5月1日,美国FDA批准Arvinas公司与其合作伙伴辉瑞联合开发的VEPPANU(vepdegestrant,ARV-471)上市,用于治疗雌激素受体阳性(ER+)、人表皮生长因子受体2阴性(HER2-)且携带雌激素受体1(ESR1)突变的晚期或转移性乳腺癌成人患者。
据公司新闻稿,这些患者需经FDA授权的诊断检测确认存在ESR1突变,且在至少一线内分泌治疗后出现疾病进展。
“此次批准不仅比原定PDUFA目标日期(2026年6月5日)提前,更开辟了全新的治疗纪元。VEPPANU作为全球首个获FDA批准的PROTAC(蛋白降解靶向嵌合体)药物,同时也是首个获批的异源双功能蛋白降解剂,标志着靶向蛋白降解技术从概念验证迈入临床应用。PROTAC作为蛋白质降解领域的革命性新概念,其里程碑意义不言而喻。”FDA前资深审评员、行业资深专家Mann Fung博士接受研发客采访时说。
FDA前资深审评员、礼邦制药首席科学官肖申博士认为:“此次获批从临床疗效来讲不是一个Game Changer。作为一个新的治疗途径,该药PFS提高有限,OS还不成熟。但技术方面很有价值,通过蛋白降解而不是依赖活性位点的抑制发挥作用。打破了以前认为的不可成药概念,这是一个全新技术路线的认证。”
从肿瘤临床医生的角度,复旦大学附属肿瘤医院主任医师、中国临床肿瘤学会(CSCO)乳腺癌专家委员会委员张剑教授对研发客评价认为:“VEPPANU的获批有益补充了ESR1突变、ER+/HER2-晚期乳腺癌在内分泌耐药后的治疗手段,开启了PROTAC这一全新药物模式在肿瘤治疗中的临床应用。作为首个口服ER降解剂,其在精准生物标志物指导下的疗效和安全性以及口服的给药方式,为内分泌耐药乳腺癌患者的治疗带来了实质性进步。”

来源|Arvinas 公司官方新闻稿(https://ir.arvinas.com/news-releases/news-release-details/arvinas-announces-fda-approval-veppanu-vepdegestrant-treatment)
商业化战略与合作伙伴选择
Arvinas与辉瑞于2021年7月宣布达成全球合作,共同开发和商业化vepdegestrant,双方分摊全球开发成本、商业化费用及利润。2025年9月,两家公司宣布计划共同遴选第三方合作伙伴,以最大化VEPPANU的商业潜力。目前,双方正按计划推进第三方合作伙伴的遴选工作,并有望尽快公布选定结果,以期尽早将这一创新疗法带给全球患者。

来源|Arvinas 公司官网 (https://www.arvinas.com/about-us/leadership/)
Arvinas总裁兼首席执行官Randy Teel博士在新闻稿中表示:“ FDA的批准对Arvinas而言是一个变革性的时刻。我们实现了首个获批药物,也是首个获批的PROTAC疗法,这基于我们自2013年以来开创并引领的技术。我们尤其振奋于在PDUFA日期之前获得批准,这体现了监管机构对这一创新疗法的认可。”
Arvinas首席医学官Noah Berkowitz博士指出:“VEPPANU的批准对患者、照护者和医生是一个重要里程碑。它为在初始治疗后进展的这种侵袭性乳腺癌患者解决了未满足的需求。今天的批准提供了一种新的口服治疗选择,与目前需要肌肉注射的标准治疗fulvestrant相比,显示出改善的无进展生存期。”
Arvinas公司背景与研发管线
Arvinas是一家临床阶段的生物技术公司,致力于通过其PROTAC蛋白降解平台改善遭受衰弱性和危及生命的疾病患者的生活。该公司基于耶鲁大学Craig Crews教授的开创性研究创立,Crews教授是首篇PROTAC蛋白降解剂论文的共同作者。通过利用人体天然的蛋白质处理系统,Arvinas的PROTAC疗法能够有选择性地高效降解并清除致病蛋白。
根据公司官网,除已获批的VEPPANU外,Arvinas拥有多个处于临床开发阶段的候选药物,包括ARV-102(靶向LRRK2,用于神经退行性疾病)、ARV-806(靶向KRAS G12D,用于胰腺癌、结直肠癌和非小细胞肺癌等突变癌症)、ARV-393(靶向BCL6,用于复发/难治性非霍奇金淋巴瘤)以及ARV-027(靶向骨骼肌中多聚谷氨酰胺扩增的雄激素受体,polyQ-AR)。公司总部位于康涅狄格州New Haven市。
内分泌耐药乳腺癌的治疗空白
张剑教授说,乳腺癌是全球女性最常见恶性肿瘤,ER+/HER2- 是乳腺癌中最常见的亚型,复发转移后的一线标准为内分泌治疗联合CDK4/6抑制剂,但40%–50%的患者会获得ESR1突变,导致耐药及预后不良。这些患者一线失败后进展快、二线选择少;CDK4/6抑制剂延长AI暴露可能增加突变率,VERITAC-2试验中43%检出ESR1突变,提示该人群存在庞大未满足的临床需求。
Sarah Cannon研究所晚期开发首席医学官兼乳腺癌研究主任、VERITAC-2试验主要研究者Erika Hamilton医学博士指出:“对于患有ESR1突变、ER+/HER2-晚期乳腺癌的患者而言,一旦标准治疗失效,二线治疗选择非常匮乏。VEPPANU的批准为为急需更多选择的患者重新燃起了希望。”
VERITAC-2试验生物标志物驱动的精准治疗
根据公司信息,此次批准基于关键性III期VERITAC-2临床试验(NCT05654623)的数据。该研究是一项全球性、随机、开放标签试验,在25个国家的213个研究中心入组了624例既往接受过CDK4/6抑制剂联合内分泌治疗的ER+/HER2-晚期或转移性乳腺癌患者,其中270例携带ESR1突变。
患者按1:1随机分配,分别接受每日一次口服200 mg vepdegestrant(28天连续给药周期)或肌肉注射fulvestrant(第1周期第1天和第15天给药,后续周期第1天给药)。试验以盲法独立中心评估的无进展生存期(PFS)作为主要终点,在ESR1突变人群和意向治疗(ITT)人群中进行评估;次要终点包括总生存期(OS)、临床获益率(CBR)、客观缓解率(ORR)及安全性。VERITAC 的临床设计见下图:
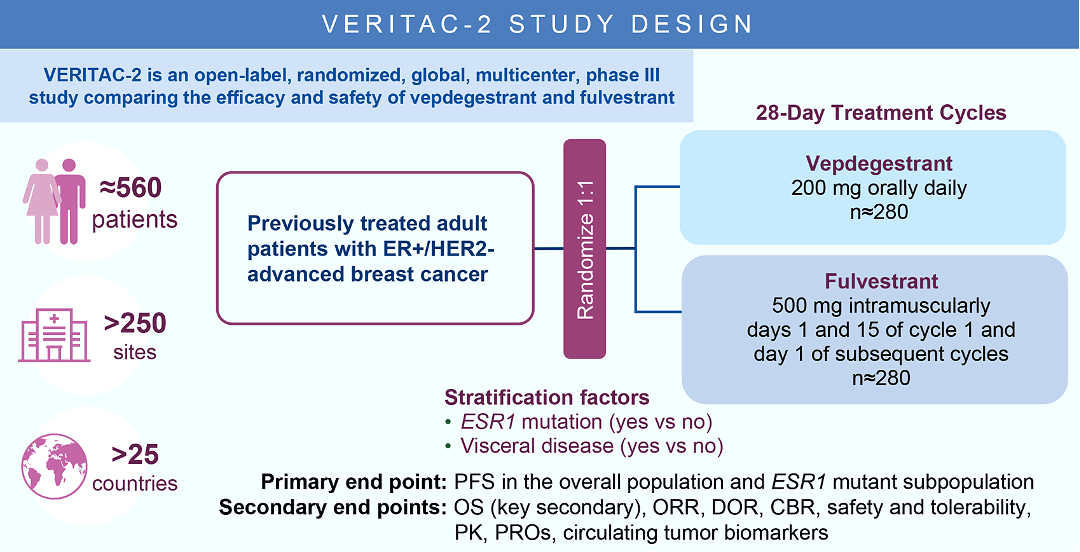
来源|https://www.tandfonline.com
在预设的ESR1突变亚组(n=270)中,vepdegestrant展现出统计学显著且具有临床意义的PFS改善。截至分析时,vepdegestrant组(n=136)的中位随访时间为7.4个月,fulvestrant组(n=134)为6.0个月。Vepdegestrant组的中位PFS达到5.0个月(95%置信区间:3.7-7.4),优于fulvestrant组的2.1个月(95%置信区间:1.9-3.5),风险比(HR)为0.57(95%置信区间:0.42-0.77;双侧P<0.001),疾病进展或死亡风险降低43%。6个月PFS率分别为45.2%(95%置信区间:36.1%-53.9%)和22.7%(95%置信区间:15.1%-31.2%)。在可测量病灶的ESR1突变患者中,vepdegestrant组的客观缓解率(ORR)为19%(95%置信区间:12%-27%),而fulvestrant组仅为4%(95%置信区间:1.6%-10%)。临床获益率方面,vepdegestrant组为42.1%(n=121),fulvestrant组为20.2%(n=119;比值比2.88;95%置信区间:1.57-5.39;P<0.001)。总生存期数据在PFS分析时仍不成熟,仅观察到16%的死亡事件。
然而,在包含无论是否存在ESR1突变的整体ITT人群(n=624)中,vepdegestrant组(n=313)的中位PFS为3.7个月(95%置信区间:3.6-5.3),fulvestrant组(n=311)为3.6个月(95%置信区间:2.2-3.8),HR为0.83(95%置信区间:0.68-1.02;双侧P=0.07),未能达到统计学显著性。
“这一结果进一步强化了ESR1突变状态作为预测性生物标志物的关键价值,而非单纯的预后标志物,显示了基于分子特征进行患者筛选和精准治疗的重要性。”肖申说。
该试验数据已于2025年美国ASCO年会上公布,并同步发表于《新英格兰医学杂志》。
公开资料显示,VEPPANU的安全性特征总体令人鼓舞。大多数不良事件为低级别(1-2级)。在vepdegestrant组中,任何级别的治疗期间不良事件(TEAE)发生率为87%,fulvestrant组为81%。3级及以上TEAE发生率分别为23.4%和约18%;严重TEAE发生率分别为10%和9%。因不良事件导致的治疗中断率极低,vepdegestrant组为2.9%(约3%),fulvestrant组为1%;vepdegestrant组因TEAE导致的剂量降低率仅为2%。任何级别的治疗相关不良事件(TRAE)在vepdegestrant组为57%,fulvestrant组为40%;3级及以上TRAE分别为8%和3%。试验期间未发生治疗相关死亡。
FDA批准的说明书包含对QTc间期延长和胚胎-胎儿毒性的警告与注意事项。
从抑制到降解的范式转变
据Mann Fung博士介绍,VEPPANU代表了现有内分泌治疗在作用机制上的根本性突破。作为一种口服生物利用度良好的PROTAC雌激素受体降解剂,vepdegestrant通过招募细胞内泛素-蛋白酶体系统,实现对雌激素受体蛋白的选择性降解和清除(见下图)。
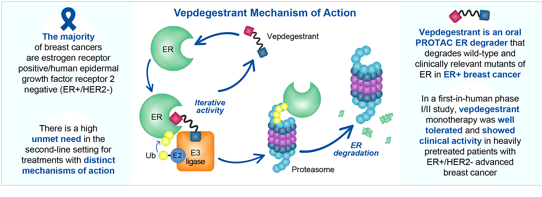
来源|https://www.tandfonline.com
“这与传统的选择性雌激素受体降解剂(SERD)如fulvestrant形成鲜明对比,fulvestrant仅能实现部分ER降解,且需要通过肌肉注射给药。ESR1突变是获得性内分泌耐药的常见机制,在二线治疗环境中约见于40%的患者,主要由CDK4/6抑制剂联合内分泌治疗的选择压力所诱导,可削弱大多数现有内分泌药物的活性。VEPPANU通过直接降解ER蛋白,靶向驱动耐药的关键生物学通路,为克服ESR1驱动的内分泌耐药提供了全新策略。”他说,这一点,非常值得国内开发PROTAC的同行借鉴。
目前,恒瑞医药、百济神州、石药集团、中国生物制药、海创药业都纷纷布局了PROTAC技术平台。“中国制药公司正加速追赶PROTAC这一新兴技术浪潮,此次FDA的批准也让我们看到了PROTAC光明的前景。”濠麦科技创始人龚元香女士说。
用药方案与伴随诊断
FDA批准的推荐剂量为200 mg,每日一次口服,随餐服用,持续给药直至疾病进展或出现不可接受的毒性。患者必须采用FDA授权的诊断检测确认ESR1突变状态后方可接受治疗。伴随此次药物批准,FDA同时批准了Guardant360 CDx作为伴随诊断工具,用于识别携带ESR1突变的适宜患者。
此前的I/II期VERITAC研究(NCT04072952)已确立了200 mg每日一次口服剂量作为推荐III期剂量。FDA曾于2024年2月基于vepdegestrant在解决严重未满足医疗需求方面的潜力,授予其快速通道资格认定。