6月29日,行业分析机构BioPharma APAC发文指出,在《生物安全法》落地、数据出境收紧及FDA对中国单中心数据日益警惕的多重压力下,全球药企在亚太地区的临床试验布局正经历根本性重构——“在中国做试验”已从商业决策演变为风险管理命题。
文章最后得出结论,在亚太地区的临床试验地选择上,找不出第二个中国,只能组合布局。
截图来源|BioPharma APAC
据报道,庞大的、未经治疗的患者群体、大刀阔斧的监管改革以及仅为西方水平一小部分的成本,使中国成为了亚太地区患者招募的首选之地。到2024年,这一引擎已不容忽视。世界卫生组织国际临床试验注册平台数据显示,中国新增药物试验超过7100项,而美国约为6000项;Citeline的年度统计显示,江苏恒瑞首次取代阿斯利康,成为全球最大的单一试验申办方。
然而,一系列政策壁垒,让人们意识到“就在中国做”已不再是一个全球申办方可以依赖的策略。2025年12月18日,《生物安全法》作为《2026财年国防授权法案》的一部分签署成为法律。该法案禁止联邦机构、受资助者和贷款接受者使用来自指定“生物技术公司关注名单”企业的设备或服务,这将影响到预计五分之四与某家中国CDMO有合约的生物制药公司。
然而,重塑亚太地区研发布局的问题不在于中国是否可行,而在于任何申办方能承受将多大比例的项目留在中国,以及哪些市场将承接转移出来的体量。
文章表示,坦白说,没有哪个国家能够取代中国,每个可行的替代方案在解决一个问题的同时,也会引入另一个问题。
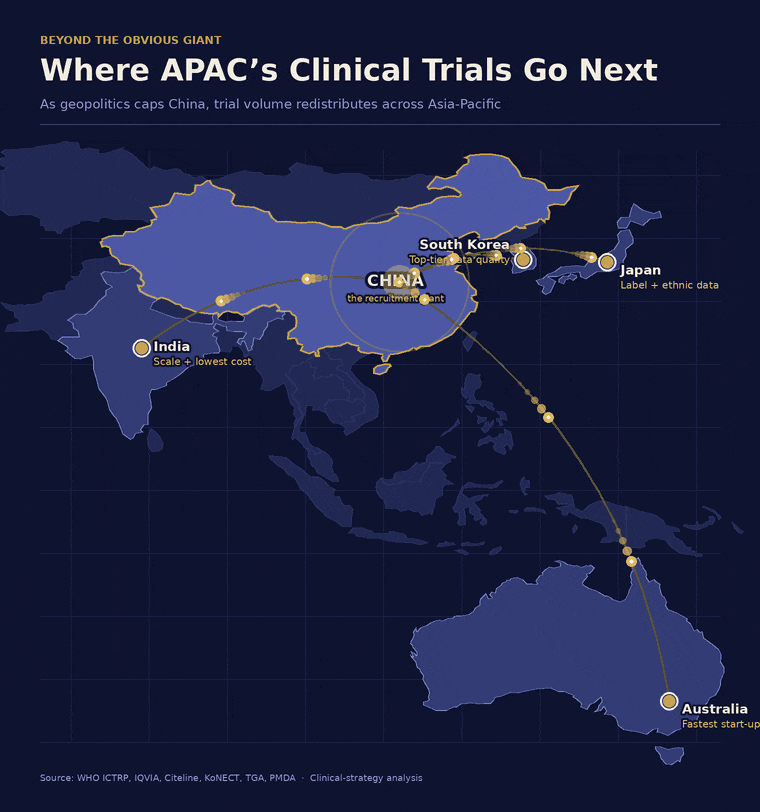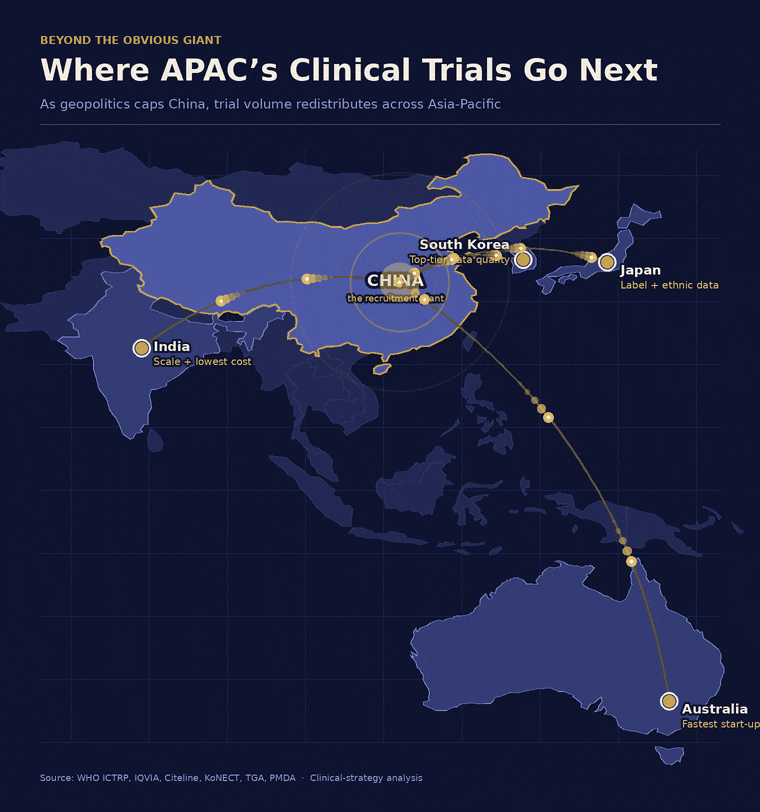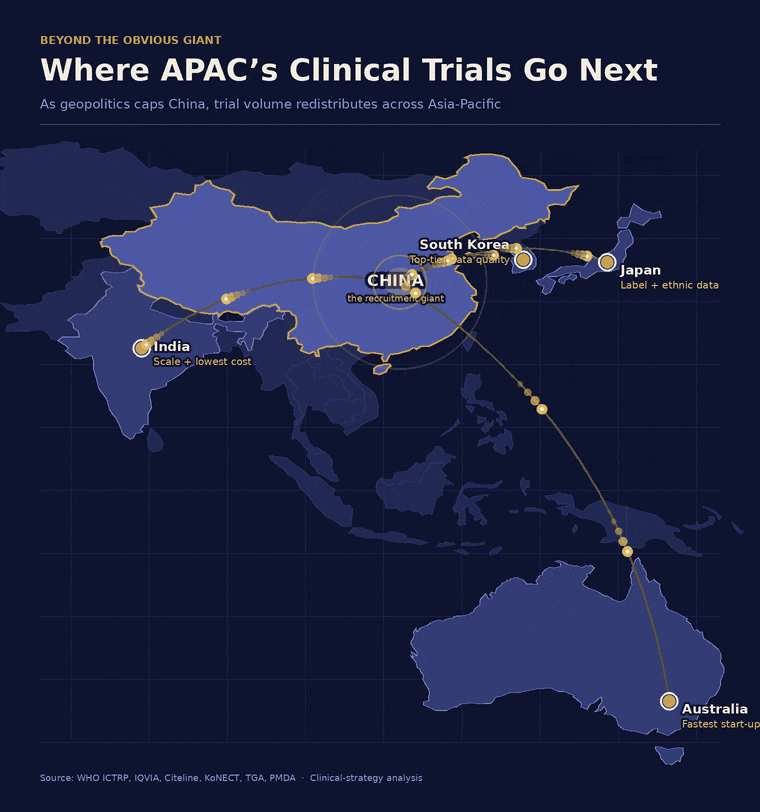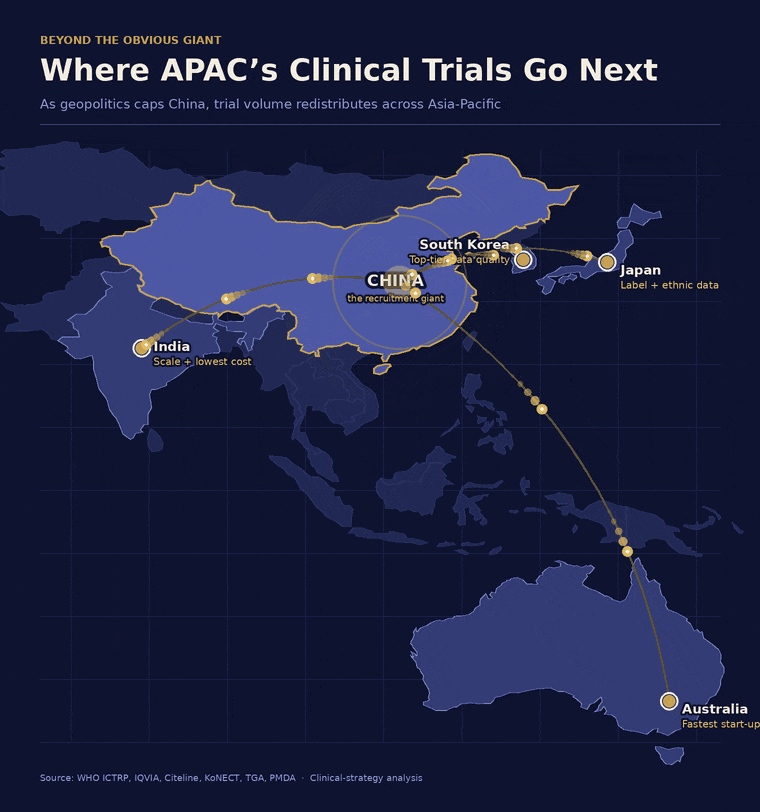
具体来说,印度能够以最低的成本提供规模优势,但其招募速度会考验申办方的耐心。韩国可以说是该地区数据质量最高的国家,但其患者基数小且竞争激烈。澳大利亚启动临床试验的速度几乎是全球最快的,但其人口上限决定了它只能专注于早期临床试验,而非追求试验规模。而日本——仍然是一个真正的创新强国——却在运营团队最看重的一个指标上不断失分:实际启动的试验数量。
亚太地区权衡取舍一览表
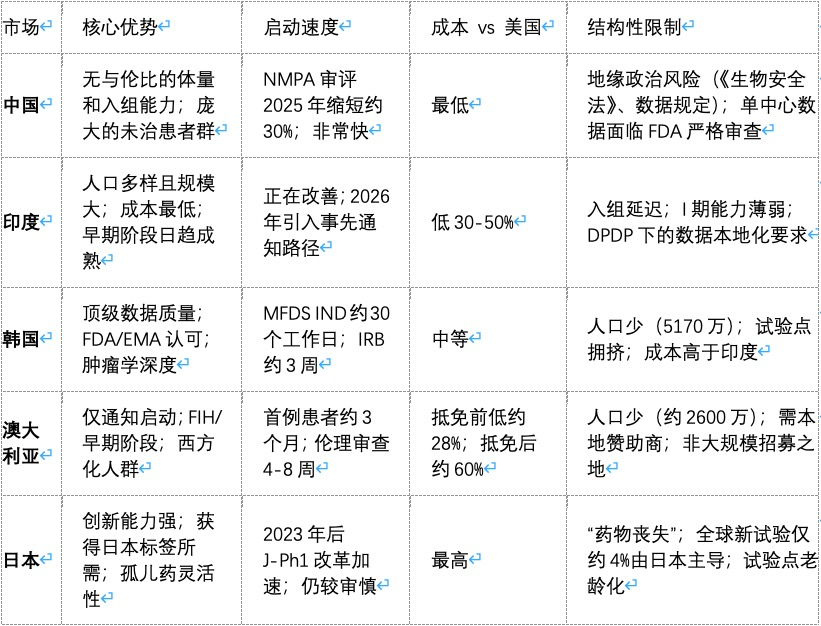
表格来源|BioPharma APAC
文章指出,寻找一个单一的替代市场是一种战略错误,因为这样的市场不存在。中国在试验速度、成本和患者数量上的组合优势,全球独一份。所以大多数项目还是会留一部分在中国做,但不会再像以前那样把鸡蛋都放一个篮子里——放多少,得看自家能承受多大的政治和监管风险。剩下的部分,就得有策略地分到别处去:
-
时间不紧张的话,去印度能省钱、能招到大量患者; -
想让监管机构挑不出毛病,去韩国,他们的数据公认干净; -
想快速起步、先验证药安不安全,去澳大利亚,启动最快; -
如果将来想把药卖进日本,那才需要考虑去日本做,因为人家要看本土数据。
拓展阅读
CDE发布《中国新药注册临床试验进展年度报告(2025年)》