In 2024, under the leadership of Dr. Yasuhiro Fujiwara, Chief Executive of the Japan's Pharmaceuticals and Medical Devices Agency (PMDA), the agency celebrated its 20th anniversary.With the Associate Executive Director,Dr. Yoshiaki Uyama's leadership, PMDA has emerged as a global leader in regulatory science. His visionary approach has positioned the country’s key medical regulatory body at the forefront of safe medical innovation.
By Donglei Mao
As the Associate Executive Director, Dr. Yoshiaki Uyama has been an integral part of the PMDA's remarkable journey. Throughout his two-decade tenure, Dr. Uyama has guided PMDA through significant progress, innovation, and a commitment to public health. His leadership has been instrumental in the PMDA's achievements, reflecting his dedication to scientific rigor and a vision for a safer, more efficient healthcare system.

Dr. Yoshiaki Uyama, Associate Executive Director of PMDA
Real-World Data (RWD) and Real-World Evidence (RWE) in Japan: Trends, Challenges, and Opportunities
Dr. Uyama is a pioneer in the integration of real-world data (RWD) and real-world evidence (RWE) into regulatory decision-making. He has been closely monitoring global advancements in this field. Recently, he expressed great interest in the innovative approach taken by the Hainan Boao Lecheng Pilot Zone in China, where RWE from overseas-approved drugs has been used to accelerate registration in China.
Since 2019, 36 international innovative drugs and medical devices have undergone RWE research there, with 15 gaining Chinese registration based on Lecheng's data. This approach has significantly shortened market access timeframes and set a precedent for global regulatory science advancement. "The Lecheng Pilot Zone is a remarkable example of how RWE can bridge gaps in drug accessibility and regulatory science," remarked Dr. Uyama.
In Japan, he has led efforts to integrate RWD and RWE into regulatory decision-making, particularly in drug development and post-marketing surveillance. He shared insights with us from the PMDA perspective on the use of RWD and RWE as external controls, highlighting both the progress and challenges in this evolving field. "In Japan, RWD/RWE has been most effectively used in areas where conducting randomized controlled trials (RCTs) is challenging, such as orphan diseases and pediatric populations," he explained. "For example, drugs like Lonafarnib for Hutchinson-Gilford Progeria Syndrome (HGPS) and processing-deficient progeroid laminopathies (PDPL), as well as Trastuzumab deruxtecan (a genetically recombinant drug) for unresectable advanced or recurrent HER2 (ERBB2) mutation-positive non-small cell lung cancer (NSCLC) that has progressed following chemotherapy, were approved based on RWD/RWE as external controls. These cases demonstrate how RWD/RWE can provide additional scientific evidence to support regulatory review."
Eight examples of RWD/RWE utilization as an EC for drug approval in Japan
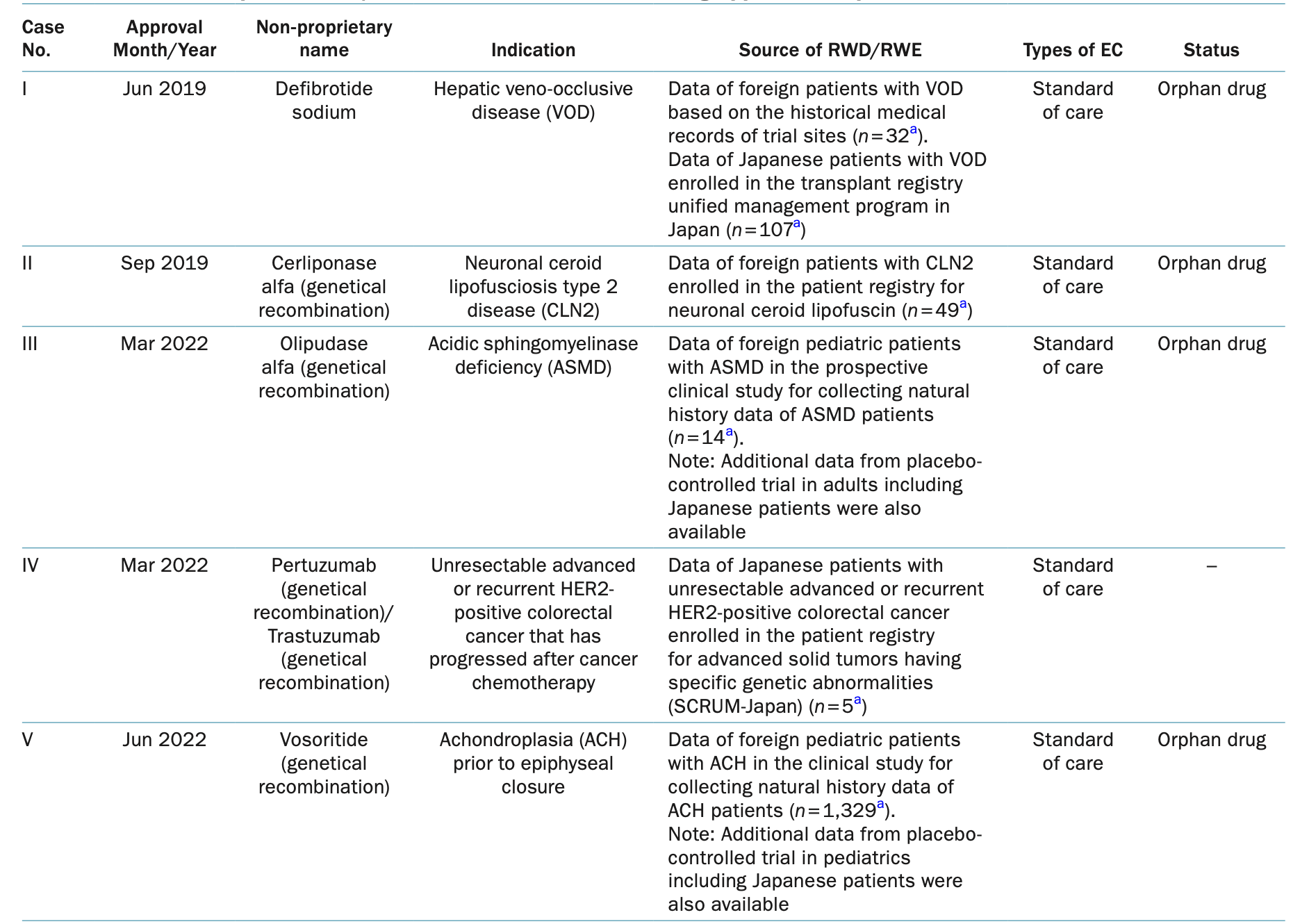
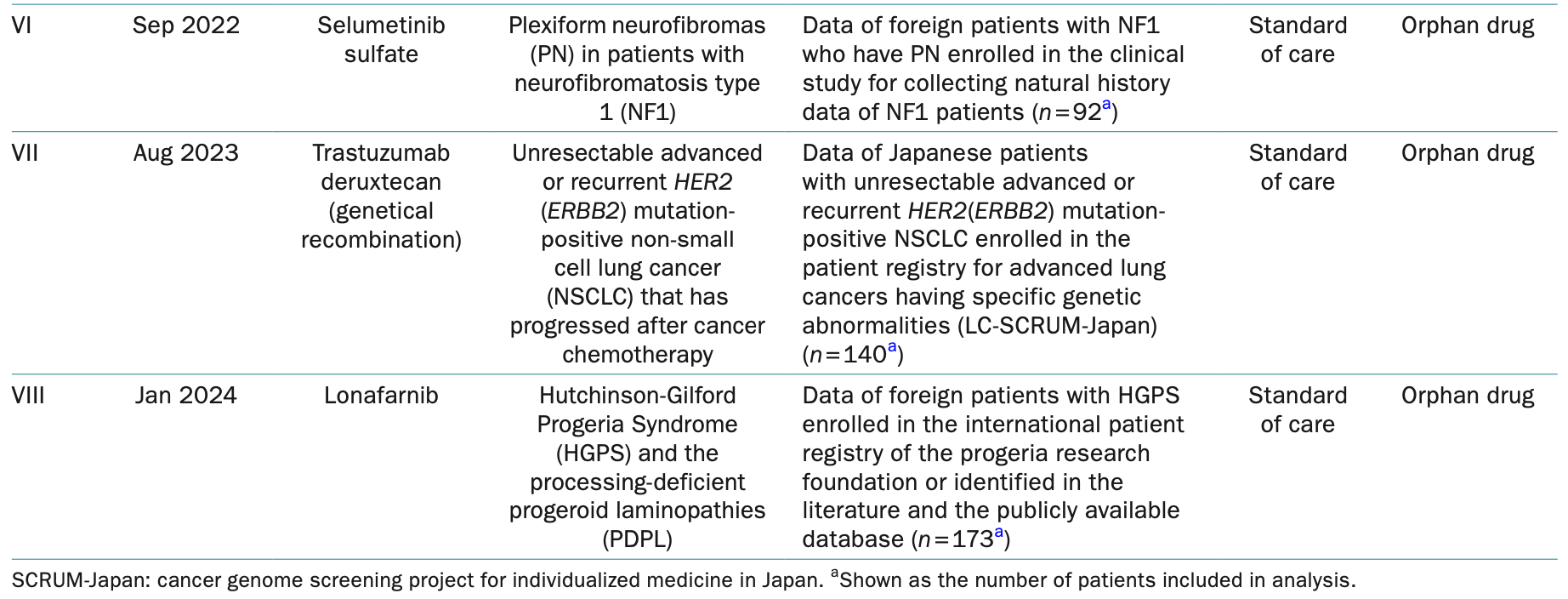
Source: 《PMDA Perspective on Use of Real-World Data and Real-World Evidence as an External Control: Recent Examples and Considerations as followed》Junichi Asano, Hiromi Sugano, Hiroyuki Murakami, Atsushi Noguchi, Yuki Ando and Yoshiaki Uyama, Clin Pharmacol Ther. 2025. DOI: 10.1002/cpt.3540:
However, Dr. Uyama also acknowledged the challenges associated with using RWD and RWE as external controls. "One common limitation is the small sample size of RWD available for orphan diseases, which makes it difficult to adjust for confounding factors and compare evidence with clinical trial data," he said. "To address this, we need to establish robust registries that intensively accumulate RWD for rare diseases and explore innovative clinical trial designs, such as Bayesian approaches."
Dr. Uyama outlined several critical points that must be considered when utilizing RWD and RWE in drug development. "A common understanding between the PMDA and sponsors about the concept and strategy of RWD/RWE utilization is essential," he emphasized. "We encourage sponsors to engage in continuous communication with the PMDA from the early stages of drug development. This includes discussing limitations, compliance with privacy protection regulations, and plans for the clinical data package." He also noted the importance of feasibility studies to understand the number of patients needed for comparisons and to identify limitations, while avoiding data dredging and maintaining scientific integrity.
Dr. Uyama further highlighted the growing importance of RWD and RWE in post-marketing surveillance. "RWD/RWE plays a critical role in monitoring the safety and efficacy of drugs after they enter the market," he said. "By analyzing real-world data, we can identify potential adverse events, update drug labels, and take proactive measures to ensure patient safety." He also pointed to the Medical Information Database Network (MID-NET) as a key tool for leveraging RWD in post-marketing activities.
Expressing optimism about the future of RWD and RWE in regulatory science, Dr. Uyama said, "The utilization of RWD/RWE is still evolving, and we are continuously learning from our experiences. As we accumulate more regulatory experience, we will be better equipped to make informed decisions and contribute to global advancements in this field." He also emphasized the importance of international collaboration, noting that initiatives like the Hainan Boao Lecheng Pilot Zone demonstrate the potential of RWD and RWE to transform drug development and regulatory processes worldwide.
Bridging Post-Marketing Safety Measures Through MID-NET
One of Dr. Uyama’s most significant contributions has been the development and implementation of the Medical Information Database Network (MID-NET), a revolutionary system that has transformed Japan’s approach to pharmacovigilance and post-marketing surveillance. MID-NET collects and analyzes real-world data from electronic health records (EHRs), claims data, and other healthcare databases, enabling the PMDA to monitor drug safety with unprecedented precision.
"The ultimate goal of our work at PMDA is to ensure that patients can use pharmaceuticals and medical devices with confidence," Dr. Uyama stated. "This requires a seamless connection between monitoring safety, responding to adverse events, and promoting proper use of a drug in clinical practice. MID-NET is the backbone that enables us to achieve this."
Under Dr. Uyama’s leadership, MID-NET has become a cornerstone of Japan’s post-marketing safety measures. By identifying safety signals that may not have been detected during clinical trials, MID-NET empowers the PMDA to take proactive measures, such as updating drug labels or issuing safety alerts, before adverse events escalate. "Real-world data allows us to anticipate potential issues and take action before they impact public health," he explained.
A Pivotal Figure at the PMDA for Over 20 Years
In 2004, PMDA was established as an independent regulatory agency, consolidating the expertise of several predecessor organizations: the Pharmaceuticals and Medical Devices Evaluation Center (PMDEC), the Organization for Pharmaceutical Safety and Research (OPSR), and the Japan Association for the Advancement of Medical Equipment (JAAME). This marked a pivotal shift in Japan's medical product oversight, promising streamlined reviews, strengthened post-market surveillance, and a steadfast focus on safety. At the time, few foresaw that Dr. Uyama would soon emerge as a key architect in shaping PMDA's future direction and initiatives.
He joined the Ministry of Health and Welfare in 1998 and has worked for the PMDA since its inception. His expertise in benefit-risk assessment, pharmacoepidemiology, and his active involvement in international harmonization efforts through the ICH made him a natural fit for the role. Over the years, he ascended the ranks, driving the agency's growth and transformation.
From 2010 to 2015 the PMDA introduced game-changing initiatives to expedite the review and approval of innovative medical products. These efforts did much to solve the country’s long-persistent problem of so-called “drug lag”. The "Sakigake" designation system, launched in 2010, was a way of fast-tracking groundbreaking therapies for patients in need. Dr. Uyama played a pivotal role in resolving Japan's drug lag issue. He spearheaded training programs to upskill the PMDA's reviewers and led the development of key guidelines, including the general principles for new drug review and the ICH E17 guideline on multi-regional clinical trials.
He also championed the use of RWD through the MID-NET system, which enhanced pharmacovigilance and post-marketing surveillance. He also advocated for increased patient involvement in the regulatory process, ensuring that the voices of those who matter most were heard.
In 2024, under the leadership of Dr. Yasuhiro Fujiwara, Chief Executive of the PMDA, the agency celebrated its 20th anniversary. Dr. Uyama reflected on the journey with a sense of pride and accomplishment. As a visiting professor at Nagoya University. At Nagoya City University and Chiba University, Dr. Uyama has also imparted his knowledge and experience to the next generation of regulatory scientists.
Dr. Uyama witnessed firsthand the transformation of the PMDA into a digital, transparent, and highly efficient regulatory body. Yet he remains clear-eyed about the challenges that lie ahead. Innovative therapies like mRNA vaccines and gene editing, for instance, place significant pressure on regulatory bodies.
"The pharmaceutical industry is advancing at an unprecedented pace, and regulators must keep up with these innovations to ensure they can effectively evaluate and guide the development of novel therapies, "he admits. "Technologies like mRNA and gene editing are transforming medicine, but they also require regulators to continuously learn and adapt to new scientific paradigms."
Looking ahead, Dr. Uyama emphasizes the importance of building a culture of continuous learning within the PMDA. "Our reviewers must stay ahead of the curve, understanding the science behind these cutting-edge therapies so they can provide clear guidance to the industry and ensure the safety and efficacy of these products," he explained. "It’s not enough to simply react to new trends; we must proactively engage with the science, collaborate with experts, and develop frameworks that support innovation while safeguarding public health."
This commitment to learning and adaptation is critical as the agency navigates the complexities of novel drug development. "The challenge lies in balancing the need for speed in bringing breakthrough therapies to patients with the responsibility to ensure rigorous evaluation," he noted. "By staying informed and adaptable, we can steer the new wave of innovation in a way that benefits patients and advances public health."
Through his leadership, Dr. Uyama is ensuring that PMDA remains not just a regulator, but a partner in the advancement of medical science, ready to embrace the future while upholding the highest standards of safety and efficacy.
His journey, intertwined with the milestones of PMDA, is a testament to the power of passion, dedication, and a relentless pursuit of excellence in ensuring the safety and efficacy of pharmaceuticals and medical devices for patients in Japan and beyond.

From left to right: Jin Chengzhe from Tigermed, Dr. Yoshiaki Uyama, and Mao Donglei from PharmaDJ.
The Past 20 Years of PMDA: A Journey of Innovation and Global Leadership
Establishment and Early Years (2004-2010)
The PMDA was founded in 2004 as an independent administrative institution. During its early years, the agency focused on:
2005: Launching operations to improve the review process for pharmaceuticals and medical devices, enhancing post-marketing surveillance, and ensuring the safety of medical products.
Enhancing Regulatory Frameworks (2010-2015)
During this period, PMDA focused on streamlining regulatory processes to support the development and approval of innovative medical products. Key initiatives included:
2010: Introduction of the “Sakigake” designation system to expedite the review and approval of groundbreaking therapies.
2012: Implementation of the Priority Review system to accelerate the approval of drugs addressing unmet medical needs.
2014: Establishment of the Consultation Service for Innovative Medical Products to provide early-stage guidance to developers, fostering innovation in the pharmaceutical and medical device sectors.
Strengthening Global Collaboration (2015-2020)
The PMDA expanded its global presence and harmonized regulatory standards through international partnerships and capacity-building initiatives:
2015: The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) was founded to strengthen its role in global regulatory harmonization, and PMDA became one of the founding regulatory members since its establishment (Note: A Japanese regulator was one of the steering committee members since 1990).
2016: Launch of the PMDA-Asia Training Center (PMDA-ATC) to provide regulatory training and support for Asian countries, enhancing regional regulatory capabilities.
2018: Introduction of the Regulatory Science Strategy to integrate advanced science and technology into regulatory decision-making, ensuring that PMDA remained at the forefront of innovation.
Advancing Digital Transformation and Real-World Data Utilization (2020-Present)
In recent years, the PMDA has embraced digital transformation and the use of RWD to enhance regulatory efficiency and decision-making:
2020: Accelerated digital transformation initiatives, including the adoption of electronic submission systems
2021: Expanded utilization of RWD through the MID-NET system for pharmacovigilance and post-marketing surveillance, improving the benefit/risk balance of a drug.
2022: Introduction of the Digital Transformation Strategy to further enhance the transparency, efficiency, and reliability of regulatory processes.
Promoting Patient-Centric Approaches (Ongoing)
PMDA has increasingly prioritized patient involvement and transparency in its regulatory activities:
Patient Involvement: Actively engaging patients in the regulatory process through public consultations and patient-focused drug development initiatives.
Transparency and Communication: Enhancing stakeholder communication through regular updates, public meetings, and the publication of review reports, fostering trust and collaboration.
COVID-19 Response (2020-2023)
During the COVID-19 pandemic, PMDA demonstrated its agility and commitment to public health by ensuring the rapid availability of vaccines and treatments:
Emergency Approvals: Played a critical role in the rapid review and approval of COVID-19 vaccines and therapies, ensuring timely access to life-saving medical products.
Regulatory Flexibility: Implemented emergency use authorizations (EUAs) and rolling reviews to expedite the availability of COVID-19-related products, showcasing PMDA's adaptability in times of crisis.
20th Anniversary (2024)
In 2024, PMDA celebrates its 20th anniversary, marking two decades of achievements in regulatory science and public health. This milestone provides an opportunity to reflect on PMDA's contributions and set future goals to further enhance its role as a global leader in regulatory innovation and patient-centric approaches.
Heartfelt thanks to PMDA for their contribution and support to this interview.
Editor:Justin Fischer