Authors: Donglei Mao (Editor-in-Chief, PharmaDJ), Tetsuomi Takano (Editor-in-Chief, Drug R&D Expert; President and CEO, t2T Healthcare Inc.)
Seven years ago, Mao Donglei’s attending the 15th DIA Japan Annual Meeting 2018 proved to be a profoundly impactful experience, particularly when Yasuhiro Fujiwara, M.D., Ph.D., Director-General, Strategic Planning Bureau, National Cancer Center and Deputy Director of the Hospital, National Cancer Center Hospital as well as Special Adviser to the Chief Executive of PMDA, delivered an inspiring opening remarks. "Though I work in regulatory affairs today, I may someday become a patient myself, and then, I would want to participate in clinical trials," he remarked. As a journalist covering the healthcare sector, this perspective prompted profound reflection. Over the years, Mao Donglei witnessed PMDA's dedication to introducing pioneering guidelines aimed at accelerating innovation for Asian populations. What remains steadfast is PMDA's commitment to enhancing the lives of patients. This vision truly resonates across the industry, touching hearts.
Dr. Fujiwara is committed to fostering collaborations with regulatory agencies and stakeholders across Asia and globally. His goal is to deliver safe, effective, and innovative medicines to those in need with his 4F philosophy. He envisions a future where new drugs and technologies originate from Asia and are then exported globally.
PMDA's fifth five-year mid-term targets include promoting regulatory science, strengthening international collaboration, and enhancing work quality and efficiency. Achieving these goals aims to make PMDA a trusted regulatory agency both in Japan and globally.
Recent R&D support measures, such as easing orphan drug designation criteria and encouraging pediatric drug development, have increased the number of orphan drugs designated in Japan. The establishment of the Consultation Center for Pediatric and Orphan Drugs Development (CCPODD) in July 2024 supports these efforts.
Moreover, PMDA is actively engaged in global collaboration. The PMDA Washington D.C. Office cooperates with the U.S. FDA as well as industry and academia to promote access to innovative medicines since November 2024, while the PMDA Asia Office in Bangkok, established in July 2024, enhances communication with Asian regulatory agencies and other stakeholders for regulatory harmonization.
Now serving his second term as Chief Executive, Dr. Fujiwara's vision and dedication continue to drive PMDA forward. What lies ahead as his next goal?

Dr. Fujiwara, a DIA volunteer, delivered his presentation and chaired the discussion with a T-shirt.
The 22nd DIA Japan Annual Meeting 2025 was held at Tokyo Big Sight from October 19 to 21, 2025, under the leadership of the Program Chair, Dr. Fujiwara.
During the conference, the authors had the opportunity to interview Dr. Fujiwara. The following is a record of that interview with partially quoting from Dr. Fujiwara’s presentation at the 22nd DIA Japan Annual Meeting 2025.

Dr. Yasuhiro Fujiwara, Chief Executive, Pharmaceuticals and Medical Devices Agency (PMDA), Japan.
Dr. Yasuhiro Fujiwara has taken his position as Chief Executive of Pharmaceuticals and Medical Devices Agency (PMDA) since April 1, 2019.
Dr. Yasuhiro Fujiwara was previously Director-General, Strategic Planning Bureau of the National Cancer Center, and Deputy Director of the Hospital, National Cancer Center Hospital. He is a medical oncologist, specializing in breast cancer. Before joining National Cancer Center Hospital, he was Deputy Director of the Evaluation Division II of the Pharmaceuticals and Medical Devices Evaluation Center of the National Institute of Health Sciences (PMDEC, which later merged with other organizations to form PMDA in 2004) of the Ministry of Health, Labour and Welfare between 1997 –2002, making his current position as Chief Executive his second appointment. Between Jan 2011 to Feb 2013, he was Deputy Secretary General of Office of Medical Innovation, Cabinet Secretariat of Japan, and led health policy issues regarding life science.
Dr Fujiwara has authored or co-authored over 280 original articles in peer-reviewed journals including Nature Reviews Drug Discovery, Lancet Oncology, Journal of Clinical Oncology, Annals of Oncology.
Q: MHLW and PMDA have introduced several R&D support measures in recent years, such as easing orphan drug designation criteria and encouraging pediatric drug development plans alongside adult drugs. The establishment of the Consultation Center for Pediatric and Orphan Drugs Development (CCPODD) at PMDA in July 2024 further highlights these efforts. How do these initiatives specifically address drug lag and drug loss?
Dr. Fujiwara: The Health and Labor Sciences Special Research Project Team announced results from their survey on drug lag and drug loss, focusing on actual conditions and solution construction on March 31, 2025.


The classification by the project team revealed the distribution of drug shortages based on necessity: Group A with very high needs comprised 14 items, Group B with high needs included 41 items, Group C with low needs had 11 items, and Group D with no needs accounted for 12 items. Additionally, 8 items were categorized under "drug loss already resolved." This survey provides critical insights into the current landscape of drug shortages and informs strategies for addressing them effectively.
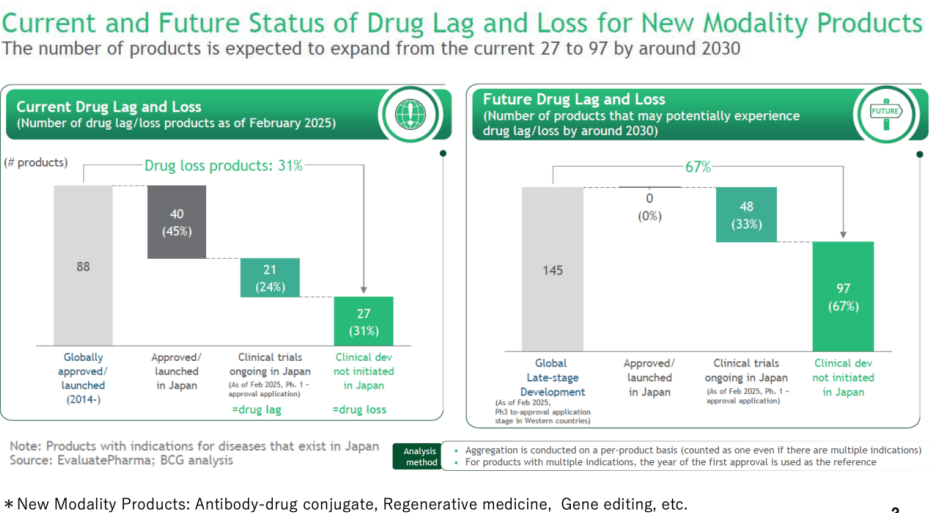
As of February 2025, there are currently 48 drug lag and drug loss products. Among the categories, 40 products (45%) are approved or launched in Japan, while 21 products (24%) are in clinical trials in Japan (Ph. 1-approval application), representing drug lag. Additionally, 27 products (31%) are in clinical development but not yet initiated in Japan, indicating drug loss. By around 2030, the number of such products indicating drug loss is projected to increase significantly from the current 27 to 97. The future projections indicate that 67% (97 products) will be in clinical development not initiated in Japan, highlighting a substantial potential increase in drug loss. Most of the items in drug lag and drug loss are oncology drugs, orphan drugs, and drugs for pediatric use.
To cope with these issues, PMDA has implemented several measures to combat drug lag and drug loss, focusing on enhancing support for orphan and pediatric drugs in Japan.

As part of these efforts, PMDA established the Consultation Center for Pediatric and Orphan Drugs Development (CCPODD) in July 2024. This center enables early and continuous dialogue between developers and regulators, helping to accelerate development timelines and reduce uncertainty. Since the establishment of this center, the number of consultations has increased. I expect that these efforts will contribute to reducing current drug lag and drug loss issues.

For orphan and pediatric drugs, PMDA addresses new policies, including earlier designation for orphan drugs and improved procedures for pediatric drug development. PMDA aims for accelerating the availability of these critical medications and promoting the clinical trial ecosystems supports this objective.

Q: Could you share recent submission and approval trends for orphan drugs, including submissions from Japan, the US, EU, and China?
Dr. Fujiwara: With the comparison between US, European Countries and Japan, it clearly shows the magnitude of the gap. Therefore, we have been placing greater emphasis on fostering development in this area.

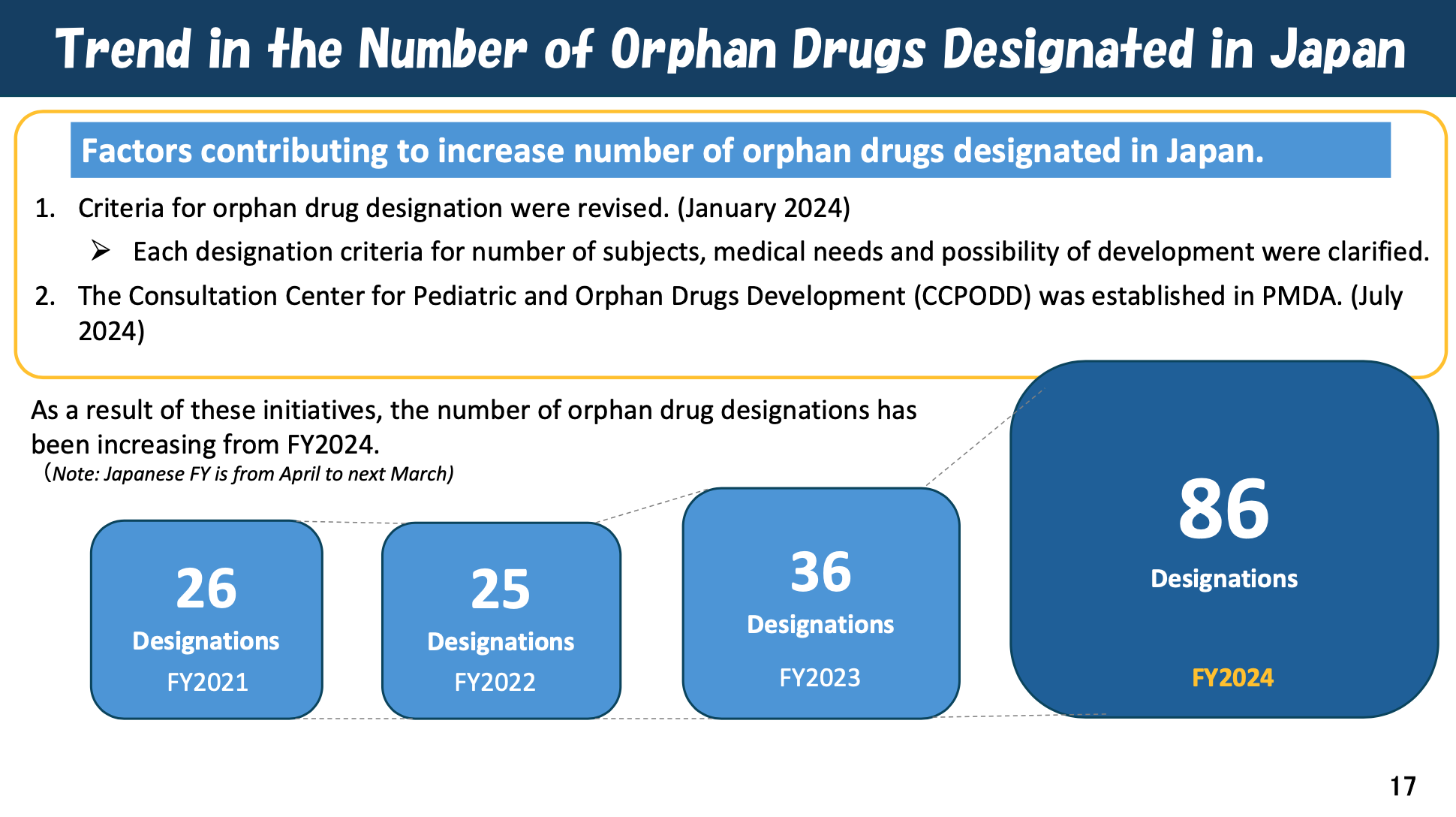
The trend in the number of orphan drugs designated in Japan reflects significant progress due to key initiatives, rising from 26 in FY2021 to 86 in FY2024. Overall, these initiatives aim to mitigate drug lag and drug loss by promoting earlier, more efficient, and more collaborative development of pediatric and orphan medicines in Japan.
Q: From 6th September 2024, Japan accepts full English NDA dossiers for foreign companies without any local affiliates or representative offices in Japan. How has the adoption of full English eCTD submissions progressed? Has this streamlined the application process for foreign sponsors?
Dr. Fujiwara: This is another method to solve current drug lag and drug loss issues, the PMDA is currently piloting the acceptance of all English submissions from companies that do not have a subsidiary or legal entity in Japan. Since many compounds developed outside Japan come from companies without Japanese subsidiaries, we must be more friendly to those companies. That's why we set up the system to accept English documents.
These kinds of initiatives will help emerging biopharma and bio-venture companies come to Japan and develop. But in the end, when the new modality is in the stage of approval, they must make Japanese documents for application to Japan. However, in the submission phase, they have English documents. If they have a Japanese subsidiary or Japanese entities, they need to translate Japanese. I think this is a really great progress for those small startup companies outside Japan to submit IND and NDA in Japan. So please come to Japan to develop. This means PMDA has always been encouraged in the industry to be more innovative.
Q: On December 25, 2023, MHLW released the “Basic principles for conducting Phase 1 studies in Japanese prior to initiating multi-regional clinical trials (MRCTs) including Japan for drugs in which early clinical development is proceeding outside Japan.” This policy allows Japan to participate in more MRCTs without the need for additional Japanese PK data. Since then, four Chinese products have successfully launched in Japan. What trends have you observed among Chinese companies regarding MRCTs that include Japan? What suggestions would you offer them?
Dr. Fujiwara: In practice, we have already accepted many MRCTs that did not initially include Japanese participants, especially in the area of orphan diseases. The new MHLW administrative notification clearly states that MRCTs without a local Phase 1 study can be accepted under certain conditions.
However, if some Chinese companies wish to submit applications of clinical trial notifications to PMDA using only Chinese data, or data from China and the U.S. combined,they need to be careful. Because it is essential for such companies to explain to PMDA the differences between clinical practices in Japan and in their own countries, especially China.
From my understanding, clinical practice in China varies greatly depending on the region. In large urban hospitals, medical standards and clinical environments are quite advanced, but for ordinary patients in less-developed areas, the standard of care can differ significantly from that of clinical trial participants. In contrast, Japan provides a very uniform level of clinical care across the population—whether a patient is wealthy or not, the access to care is consistent. This is a major difference between Japan and countries like China or even the United States, and major merit of Japan. Therefore, when Chinese companies plan to introduce their drugs to Japan, I strongly recommend they demonstrate to PMDA that the data they hold are scientifically and clinically applicable to Japanese patients. They need to justify this with appropriate explanations of extrinsic factors—such as clinical settings, diagnostic practices, and standard of care differences—rather than relying solely on intrinsic factors like genetics or metabolism, which we already known are relatively similar between Japan and China.
Any Chinese companies would realize that incorporating Japan into their development plans was feasible; as a result, utilization of the Phase I waiver has increased. When conducting MRCTs in Japan, it is essential to look for qualified physicians, clinical institutions, and a suitable CRO that can execute the trial properly. This requirement is the same as when clinical studies are carried out outside Japan essentially.
In conclusion, this new policy is very encouraging and opens doors for more efficient global drug development. However, please keep in mind that its successful application requires careful scientific justification and proactive dialogue with PMDA. Earlier discussions with our review teams can help clarify expectations and evaluate whether the available data are suitable for submission in Japan.
Q: What key areas are covered by Early Considerations, which have been published more than 20 times since 2024?
Dr.Fujiwara: The recently published Early Considerations serve as preliminary reference information and a perspective for promoting the practical application of new technologies, innovations, and the development of innovative drugs, even though scientific knowledge and information may not yet be fully accumulated.

These considerations cover several key areas, including handling Japanese data for confirming biosimilar comparability to original biopharmaceuticals, considerations for externally controlled trials, nonclinical safety matters in initial clinical trial notifications, discussions with PMDA using the ICH S1B (R1) guideline in approval applications, and points to consider in developing drugs for Pediatric Inflammatory Bowel Disease and the clinical efficacy evaluation of drugs for Palmoplantar Pustulosis.
Q: The keynote speaker from Boston on DIA Japan Annule Meeting, Dr. Shinichiro Fuse,from TPG Life Sciences Innovations highlighted that some of the recent innovations from China are quite impressive. Could you share your thoughts on Japan’s innovation capacity compared to emerging markets like China? How would you assess Japan's innovation capacity in this field compared to the US?
Dr. Fujiwara: Japan’s innovation capacity is generally strong. However, there is one major weakness in our innovation ecosystem—the academic sector often lacks the capacity to translate research outcomes into practical applications or bring them to market. This is a key challenge for Japan. Our basic research would be excellent; for example, Japan has been honored multiple Nobel Prize winners in recent years. But the translation from academia to industry—what we call “from bench to bedside”—is still limited. That is why I often emphasize that the government should increase investment in the application stage of research, to help bridge this gap between discovery and commercialization.
If you look at China, the government provides very strong support for innovation in life sciences, both financially and structurally. They have substantial funding, and many scientists who studied or worked in the United States are now returning to China, further strengthening their research ecosystem. I recognize that emerging biopharmaceutical companies (EBPs) developing new medicines exist in Japan, and I believe that their capacities are adequate. The challenge is to sustain the development of new drugs by these EBPs and successfully to translate them into market products. From my personal view, I believe that collaboration with companies outside of Japan, such as conducting MRCTs, rather than limiting development solely within Japan, would be highly beneficial.
In contrast, Japan once had many researchers and physicians conducting studies abroad—especially in the United States, at institutions like the NIH or major universities. When I was in the U.S., I met many Japanese researchers there. However, today it is becoming difficult to find such examples. Younger generations in Japan tend to stay in Japan and show less enthusiasm for studying or conducting research abroad compared to my generation.
The world is changing rapidly, and I believe young people must be encouraged to look beyond Japan—to experience other cultures, learn from international settings, and bring those perspectives back home. We need to foster a mindset where young researchers aspire not only to advance their own careers but also to make meaningful contributions to their communities and to Japan as a whole. Encouraging more international exposure and a broader worldview to young people is essential to revitalizing Japan’s innovation capacity.
Q: What were the objectives and outcomes of the inaugural meeting of the Public-Private Council for Enhancing Drug Discovery Capabilities?
Dr. Fujiwara: The inaugural meeting of the Public-Private Council for Enhancing Drug Discovery Capabilities was held on June 26, 2025, convening approximately 30 key domestic and international stakeholders.

The assembly included Prime Minister Ishiba, Chief Cabinet Secretary Hayashi, Minister of Health, Labour and Welfare Fukuoka and me , alongside representatives from pharmaceutical companies, venture capital firms, start-ups, and academia. The primary objective was to discuss initiatives aimed at improving Japan's drug discovery ecosystem and to establish a collaborative framework between the government and private sectors to advance this goal.
To elevate clinical trial infrastructure, initiatives focus on securing and developing human resources, such as Clinical Research Coordinators, biostatisticians, and data managers. Improving education about clinical trial methodology and significance, both at medical and nursing schools, is also prioritized, alongside establishing clear career paths for supporting staff and providing economic incentives like national qualifications and improved remuneration. These efforts align with strategies outlined in the 3rd Basic Plan for Science and Technology (2006-2010).
Educating the public and healthcare professionals on the importance of clinical trials is crucial. Additionally, raising awareness about clinical trial registration numbers in the pharmaceutical regulatory field is essential to prevent opaque research results and ensure transparency. This includes establishing a Japanese version of the RePORT tool by National Institute of Health (NIH) to track clinical trial topics.
Q: What key activities are PMDA undertaking to improve the clinical trial environment in Japan?
Dr. Fujiwara : PMDA is actively improving the clinical trial environment in Japan through several key activities. These include the international guideline on ICH-E6 (R3), which provides guidance on the clinical development lifecycle, considers "Quality in Clinical Trials" as "Fitness for Purpose," and introduces the concept of Quality by Design (QbD).

Additionally, PMDA is also leading a project to promote the introduction of a clinical trial ecosystem, consolidating opinions on current clinical trial environments. These efforts, are set to significantly improve the clinical trial landscape in Japan.
Q: How is PMDA enhancing reviewer capacity and expertise? For example, are there plans for recruitment or training programs focused on AI-driven reviews?
Dr. Fujiwara: PMDA recently released its Action Plan for the Use of AI in Operations at PMDA (Prepared on September 26 in Japanese and English) on October 10, which outlines our current thinking and initiatives regarding the application of AI technology in regulatory review processes.
We developed this action plan over several months, inspired by similar initiatives from the European Union and the U.S. FDA, as we believe it is important for PMDA to align with these global regulatory trends.

Since many companies are now actively utilizing AI tools in their research and regulatory documentation, PMDA also needs to integrate these technologies into our own internal review processes. We are therefore enhancing our reviewers’ capacity through training to ensure that PMDA can assess AI technologies appropriately and efficiently.
Q: How has PMDA integrated patient perspectives into the approval processes of pharmaceuticals and medical devices?
Dr. Fujiwara: PMDA has integrated patient perspectives into the approval processes of pharmaceuticals and medical devices through multiple initiatives. Launched in May 2019, the PMDA's Patient Centricity Working Group (WG) evolved into a sustained effort by October 2024. Emphasizing a core principle of "Patient First." In September 7, 2021, PMDA published its "Guidance on Patient Participation," delineating methods for involving patients in clinical development.
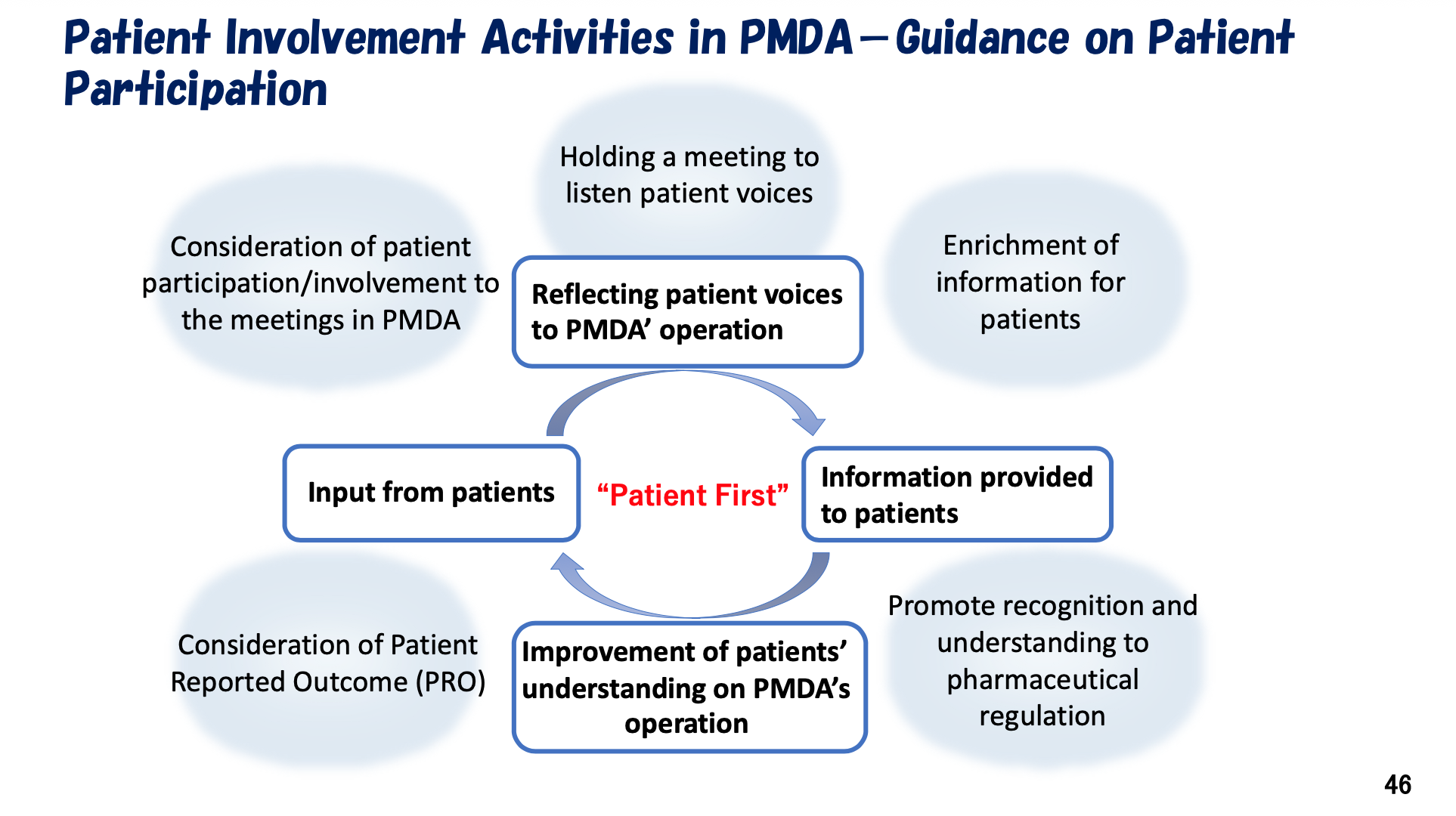
Additionally, PMDA engages in external workshops, collaborates with patient associations. Central to PMDA's activities are key processes including gathering patient input through meetings, providing relevant information to patients, and improving patient understanding of pharmaceutical regulations—all forming a framework to ensure continuous influence of patient feedback on PMDA's decisions.

Concurrently, ICH is developing the E22 guideline titled "General Considerations for Patient Preference Studies," currently at Step 1 (Concept Paper Stage). I recognize this guideline aims to optimize the use of patient preference information in drug development through a globally harmonized approach.
Through these concerted efforts, PMDA and ICH are advancing towards more patient-centered healthcare decision-making.
Q: What is the mission of the PMDA Washington D.C. Office which was established in November 2024?
Dr. Fujiwara: This involves cooperation with the U.S. FDA and related administrative agencies to promote further access to innovative medicines, medical devices, and regenerative products, as well as engaging in discussions on marketing authorizations and post-marketing measures.

Additionally, the office aims to provide more opportunities for communication with stakeholders to offer information on Japanese regulations, operating in the same time zone to minimize time-zone differences and serving as a "General Consultation Service" for start-up companies during early development stages in Japan.
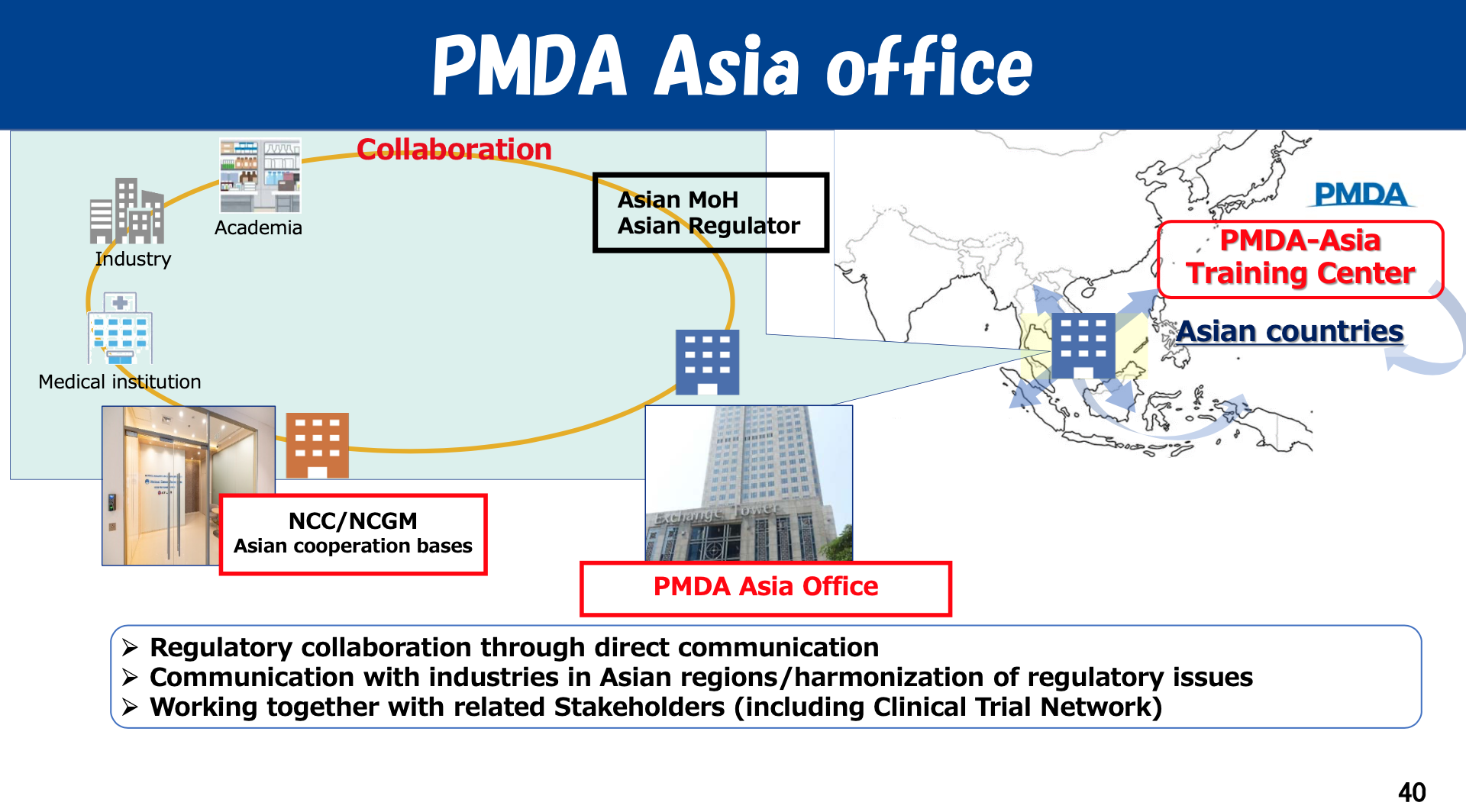
Q: With the establishment of the PMDA Asia Office in Bangkok in July 2024, what are PMDA’s strategic priorities for collaboration with Asian regulatory agencies such as the Thai FDA? How does PMDA define “reliance” in regulatory contexts?
Dr. Fujiwara: Through the Bangkok office, PMDA aims to enhance communication and information exchange with Asian regulatory agencies. The staff there are actively traveling throughout the region—especially in Southeast Asia—to meet regulatory officials in person. These face-to-face meetings are important. Meeting in person helps build mutual understanding, trust, and a clearer awareness of PMDA’s activities and regulatory philosophy.
Innovative drugs are typically approved first in advanced economies and are then submitted to other countries, such as those across Asia. Therefore, we can facilitate rapid access by enabling efficient transfer of a drug that has already been approved in Japan to other Asian countries. I believe that harmonization of regulatory differences between Japan and Asian countries is achieved through these efforts.
We understand “Reliance” means that a regulatory authority may, at its own discretion, utilize the decisions made by other authorities. Each regulatory authority bears responsibility for the efficacy, safety, and quality of drugs in its own jurisdiction.
Q: We totally agree. Face-to-face meetings truly bring people closer together. During the DIA Japan and China Annual Meetings, we discussed the collaboration among Japan, Korea, and China. About ten years ago, Dr. Tatsuya Kondo, Former Chief Executive of PMDA, also promoted mutual cooperation and potential regulatory recognition among these three major countries in Asia. What is the current status of collaboration between Japan, Korea, and China? What is the role of the PMDA Asia Office in fostering regulatory collaboration across Asian countries?
Dr. Fujiwara: Regarding expansion into East Asia, PMDA’s regional strategy has evolved from focusing mainly on Northeast Asia to now including Southeast Asia as well. However, our basic policy toward Asia remains consistent—we consider not only East Asia but the entire Asian region to be very important. With Korea and China, PMDA continues to maintain close and frequent dialogue.
We hold many meetings with both countries. At the same time, we are also broadening our engagement toward South Asia through the Bangkok office. This does not mean that we are shifting attention away from East Asia—on the contrary, Japan, Korea, and China will always remain key partners for PMDA. We have strong and positive relationships with our counterparts in both Korea and China, and we will continue to build on that foundation while expanding our network across Asia.
The PMDA's vision for the Asian region is shaped by economic growth, population growth, and an aging society, leading to a surge in EBPs that are increasingly developing new products.
Q: Our last question is about your personal career. Your presentation was truly inspiring when you mentioned that you joined PMDA because you wanted to work more closely with patients. Could you reflect on what you consider your greatest achievements from your previous term as Chief Executive, and what are your goals and expectations for your current term, which began in 2024? What motivates you, and what challenges do you foresee in the years ahead?
Dr. Fujiwara: My journey to becoming the Chief Executive of PMDA is rooted in my deep consideration for patient-centricity sentiments. Before the establishment of PMDA in 2004, I served on a committee at PMDEC, where I was tasked with recording research funds related to industry-academia relationships. During that time, I noticed a significant disconnect basic researchers showed little interest in patients, and patients remained unseen and unheard in the drug develop process. This realization fueled my determination to bridge the gap among patients, researchers, and regulatory bodies. I saw an opportunity at PMDA to make a tangible difference.
Upon my arrival, I was struck by the fact that even within PMDA, the focus was primarily on interactions with pharmaceutical companies, with little to no direct engagement with patients. This further solidified my resolve to advocate for a patient-centric approach. I began by establishing a patient working group to set a new direction. I also spearheaded the creation of guidance principles aimed at incorporating patient voices into PMDA's operations. My vision was to create a system where patients and the public could actively participate in meetings and decision-making processes, ensuring that their needs and concerns were heard and addressed.
One of my key initiatives is to listen to the voices of victims of drug-related harm, providing a platform for ordinary patients to share their experiences and opinions. I believe that by creating more opportunities for patients to engage with PMDA, the agency can better understand their needs and improve the overall healthcare system. My commitment to a Patient-First approach is evident in my efforts to encourage public participation in PMDA's various meetings and to develop a system that allows for decision-making.
My ultimate goal is for Asia to become a region recognized globally for its strong capacity in drug development. At present, most innovative drugs are still developed in the United States or Europe, and only later do large pharmaceutical companies bring them to Asia for further development or market expansion. I believe this model is not ideal for us in Asia. While China has developed significant capabilities to innovate and conduct its own research, many other Asian countries still lack sufficient capacity to develop drugs or medical devices independently.
Now is the time for us to change that. We must encourage and support Asian scientists, regulators, and companies to accelerate the development of drugs by Asian people, for Asian patients. That is my long-term vision—and the mission I see for PMDA as well. PMDA should serve not only Japan, but the entire Asian region. Countries in Asia—Japan, China, Korea, and others—share many of the same diseases and health challenges and together represent a huge population. If we can develop medicines suited to our own patients and healthcare systems, we can then bring those innovations outward—to the United States, Europe, and beyond.
Currently, large Western pharmaceutical companies often prioritize regions like Africa or South America for expansion, while Asia—except for China—is still not viewed as a top-tier market. I hope to change that perception. I often say to my colleagues in the U.S. and Europe: “Please come to Asia, collaborate with us, and develop together with Asian partners.” In the future, I want to see new drugs and medical technologies originating from Asia—developed using Asian patients’ data, and then exported globally.
That, to me, would be the real achievement. My motivation has always been to promote better healthcare—not just for Japan, but for all people across Asia. I believe every person in this region deserves access to safe, effective, and innovative medicines. That is the vision I want to advance during my current term as Chief Executive of PMDA. As Chief Executive of PMDA, it is essential to view the PMDA as a safeguard for the health and safety of the Japanese public. This mindset forms the backbone that guides PMDA’s leadership.
PMDA’s fifth five-year mid-term targets outline what we aim to achieve: ① To actively promote the practical application of regulatory science. ② To strengthen our capacity for international collaboration and proposal development. ③ To enhance the quality of our work and drive further efficiency gains. By achieving these goals, I hope the PMDA becomes a regulatory agency that is trusted not only by the Japanese public but also by the global community. I would like to realize these targets as my immediate priority.

Donglei Mao with Dr. Fujiwara,Dr. Yoshiaki Uyama, Associate Executive Director of PMDA and Naoyuki Yasuda, Special Adviser to Chief Executive and Associate Executive Director of PMDA. Donglei Mao and Tetsuomi Takano with Dr. Fujiwara and Dr. Yoshiaki Uyama
Donglei Mao and Tetsuomi Takano with Dr. Fujiwara and Dr. Yoshiaki Uyama