編集長コメント (2023年12月6日発行、第62号に寄せて)
今回の第62号では、2023年11月8-10日に上海で開催されたChinaTrials15の初日のワークショップに組み入れられた、研発客、医薬研発達人、Tigermedの共催による「中国の製薬企業向けの日本プロモーションセッション」である ” From IND to NDA: How To Implement the Clinical Development Plan in Japan” について取り上げた。
ChinaTrialsは、Lychee Groupによって2008年に創設された「中国における医薬品開発と治験」に焦点を当てた商業系最大のカンファレンスで、毎年11月、2015年の第8回までは北京で、2016年の第9回以降は上海で開催されている。なお、2020年はコロナ禍にて開催が無かったため、今回の2023年11月が第15回となっている。また、2021年の第13回以降は、Lychee Groupと研発客の共催となっている。
近年のChinaTrialsは約500名前後の参加規模であるが、特徴的なのは、中国製薬企業の経営層やグローバル製薬企業の中国R&D Executivesの登壇者が多いことと、プレゼンよりはラウンドテーブルに比重が置かれていることである。なお、DIA Chinaより規模は小さいが、ステージで話されている経営陣同士のほとんど本音の討論内容は奥が深いし、質疑応答の平均レベルも高い。
さらに、中国医薬創新促進会 (中国薬促会、China Pharmaceutical Innovation and Research Development Association, PhIRDA) 主催で、規模が巨大かつカバー範囲も広大な、中国医薬創新与投資大会 (China Biomed Innovation and Investment Conference, CBIIC) と比べると、より医薬品開発と治験に関連したセッションに絞られていることと、規模がこじんまりとしている分、ChinaTrialsでは登壇者と直接話す/知り合える/WeChat連絡先交換ができるチャンスが多いというメリットがある。
[参考: 医薬研発達人第12号(2021年12月6日発行)「第6回中国医薬創新投資大会(CBIIC):導入(Buy)、フォロー(Follow)、改良(Improve)から真のイノベーションへ」、医薬研発達人第47号(2023年4月24日発行)「2022 CBIICにて中国医薬品規制改革の成功の軌跡と今後の課題を見る」]
医薬研発達人が中国で日本プロモーションセッションを行うのは、6月18日の蘇州での第15回DIA中国年会Day 3に次いで今年2回目であるが、前回6月のフロアと違って、今回11月のフロアでは、聴衆からの大きな熱量と真剣さを感じた。
[参考: 医薬研発達人第51号(2023年7月3日発行)「第15回DIA中国年会報告(第一弾)中国バイオテク企業が日本へ進出するということ」、医薬研発達人第52号(2023年7月17日発行)「第15回DIA中国年会報告(第二弾) 医薬研発達人創刊2周年記念号」]
すなわち、具体的かつ的を射た質問の数々からも、今回11月のフロアでは、聴衆が「日本市場進出や日本での治験実施」に明らかに興味を持っていて、実際よく勉強しているように感じた。
その理由として、今年6月から11月までの5か月間の変化を振り返ると、日中両国政府の関係が少し改善した、中国の経済減速がより顕在化してきた、長引く円安、コロナ禍からの回復状況の持続などが挙げられるが、今年11月のChinaTrials15の中国人参加者 (ちなみにChinaTrials15では3日間を通じて自分以外には一人の日本人も見かけなかった) と話していて圧倒的に感じたのは、「今年6月9日に厚労省から出された有識者検討会報告書」や「今年7月や9月の薬事検討会結果」に代表される「ドラッグロス・ドラッグラグを解消するために大胆な規制緩和もいとわず真剣に立ち向かう日本国や日本政府の本気度」を中国の産業界が肌で感じ取り、これからは日本に大いにビジネスチャンスあり、と捉えていることである。
そのことがChinaTrials15以降の中国産業界の動きにも早速表れている。
先週、11月28日から3日間、PhIRDAの日本訪問団が15社30名に迫る規模で東京(&神奈川)にやってきた。主目的は、日本でのパートナー探しで、錚々たる面々が中国各地から同時に来日し東京に集結した。
私は、PhIRDAの支援者兼日中医薬品開発専門家として、初日に彼らにプレゼンを行ったほか、製薬協との会議、医療機関訪問等をサポートするなど、彼らに3日間帯同した。
このPhIRDAの日本訪問は、完全クローズの非公開イベントなので、本来、医薬研発達人には何も書くことができないが、昨日12月5日にPhIRDA自身が、WeChatで、先週の日本訪問報告を行い、公開情報となったので、今、こうして医薬研発達人でも本イベントについて記述することが可能となった。本62号の発行日が2日遅れの12/6(水)となったのは、このような事情によるものなので、お許しいただきたい。
このWeChatで公開されたPhIRDAの日本訪問報告は、製薬協や製薬企業での集合写真のほか、いつどこで誰とどんな行事を行ったのか、相当細かく書かれているので、中国語表記ではあるものの、一度ご覧になることをお勧めする。
中国药促会代表团到访日本制药工业协会及武田制药(2023年12月5日、PhIRDA発行)
今回のPhIRDA日本訪問団の熱量はハンパなく、本当に火傷しそうになるくらい熱かった。6月のDIA中国年会の頃とは全く比較にならず、11月上旬のChinaTrials15の時よりも格段にヒートアップしていた。
来年、医薬品開発/ビジネス周辺の日中連携は、産業界をドライバーとして大いに前進することを確信した。
高野 哲臣(t2T Healthcare株式会社代表取締役社長)
文 | 毛 冬蕾(Mao, Donglei)
今年6月16-19日に蘇州で開催された第15回中国DIA年会のDay 3において、“Japan as the next destination for Chinese biotech/biopharma -Demystify its hurdles/barriers”と題する日中共同セッションが行われたように、中国の製薬企業にとって、世界第3位の医薬品市場を有する日本は、近い将来、神秘的で遠い存在ではなくなるであろう(医薬研発達人第51号(2023年7月3日発行)「第15回DIA中国年次総会報告書(第一弾) 中国バイオテク企業が日本へ進出するということ」参照)。
では、中国企業が日本へ進出しようとする場合、その計画をどのように策定すればよいか。中国における初期段階の臨床データを利用して日本の治験計画を構築し、新薬開発プロセスを加速するにはどうすればよいか。日本では医薬品の治験中のファーマコビジランス (PV) についてどのような要件があるか。日本で治験をスムーズに進行させるためのコツは何か。
11月8-10日に上海で開催された第7回『研発客』臨床年次総会兼ChinaTrials15のDay 1において、”From IND to NDA: How To Implement the Clinical Development Plan in Japan”と題する日本関連セッションが、ChinaTrialsの15回の歴史の中で、初めて設けられた。このセッションは、3つの演題とパネルディスカッションによって構成された。
 『医薬研発達人』編集長の高野 哲臣氏
『医薬研発達人』編集長の高野 哲臣氏
まず、1番目のスピーカーである『医薬研発達人』編集長の高野 哲臣氏は、” An Overview of the Japanese Pharmaceutical Market and PMDAʼs Drug Regulations: From IND to NDA - How To Implement the Clinical Development Plan in Japan”と題して、日本の医薬品市場、治験、承認申請、医薬品規制等の現状や最新動向について包括的に紹介した。
同氏は、今年7月10日に開催された第1回創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会(以下、薬事検討会)で議論された「オーファン指定要件の見直し」や今年9月13日に開催された第3回薬事検討会で了承された「国際共同治験開始前の日本人PKデータ原則不要化」、ならびにそれに伴う「国際共同治験に関する基本的考え方について」、「同(参考事例)について」、「国際共同治験開始前の日本人での第I相試験の実施に関する基本的考え方について」との日本独自のMRCT guidelines (国際共同治験に関する基本的考え方 3シリーズ) の近未来の改定可能性について言及した。


高野哲臣氏の講演スライドより
日本における上記の規制変更について、筆者は中国の製薬企業数社にインタビューを行った。インタビューに応じたほとんどの企業は、今回の日本の規制変更が、日本で新薬を発売したいと考えているすべての中国企業にとって大きな朗報であると考えている。中国人と日本人は遺伝子や疾患において非常に類似しているため、PK試験に関する今回の規制変更により、中国企業や他の外国企業は、この機会を捉えて、日本でより多くの後期段階の臨床開発を実施するようになるだろう。臨床試験の一部省略は日本における開発費のコストダウンにも繋がり、日本での開発が加速されるであろう。ちなみに、Yaozhi.com の統計によると、2010 年 3 月から 2023 年 11 月までに中国のバイオ医薬品企業 (台湾を含む) が日本で実施した臨床試験は40であったが、2022年だけでもその数は9に上り、近年急速に伸びていることを示している。
日本において抗悪性腫瘍薬Gumarontinibの承認申請を行っている海和生物医薬(HaiHe Biopharma Co., Ltd) のCEO、董瑞平(Dong, Ruiping)博士は、「日本人を対象とした第1相試験の必要性については長い間議論されてきた。PMDAはすべての品目で第1相試験の実施を義務付けるわけではないが、今回の変更により、薬剤の作用機序や試験目的、適応症などに応じて事前にPMDA相談を行い、申請者が該当品目の安全性および人種上の差が小さいことをPMDAに納得させることができれば、日本で第1相試験を実施する必要はなくなるかもしれない」と述べた。
元FDAシニア審査官である思路迪医薬(3D Medicines)のチーフメディカルオフィサーである肖申(Xiao, Shen)博士は、これはFDAのアプローチに似ていると考えており、「FDAは米国人に対する安全性試験の義務付けを行っていない」と述べた。「それを見本に、新薬が日本で承認され、中国と日本の標準治療に差がなく、民族差が少ないことが示されれば、中国での承認を加速することにも役立つだろう。」現在、同社の PD-L1 抗体Envafolimabは、日本を含むグローバルでpivotal phase 2 studyを推進している。
緑葉製薬(Luye Pharma Group)の国際研究開発臨床薬理学責任者である李皛(Li, Xiao)氏は、このニュースは中国のみならず東アジア各国の企業にとっても朗報であり、日本やその他の国での同時開発計画を立案するのに役立つだろうと考えている。「中国企業が新薬開発計画を策定する際には、日本での研究開発費を再検討した上で、日本で臨床試験を積極的に行う企業が増えていくだろう」と。
実際、日本における臨床開発を強化する方向で、中国の複数の製薬企業が既に動き始めている。11月28日から30日にかけて、中国医薬創新促進会(中国薬促会、China Pharmaceutical Innovation and Research Development Association, PhIRDA)は、中国のバイオ医薬品企業の幹部ら26名による東京/神奈川訪問を行った。その中には、綠葉製薬、亞盛医薬(Ascentage Pharma)、科倫薬業(Kelun Pharma)、復宏漢霖(Henlius)などの大企業も含まれている。

中国薬促会(PhIRDA)日本訪問代表団
また、来る12月14日に、『製薬ハンターズクラブ』と『医薬経理人』の後援で中国のバイオ医薬品企業の幹部22名が大阪を訪れ、地元の大学や企業との交流を通じて日中製薬協力の新たな機会を模索することになっている。
協和キリン中国の開発部門責任者である丁锎(Ding, Kai)博士は、日本の厚労省が、pivotal MRCTに入る前に日本人を対象としたPKデータ要件を緩和することは、イノベーションを活性化する良い流れであると考えている。「新薬の種類や適応症によって、pivotal studyに入る前に、医薬品の安全性、人種差、潜在的な臨床価値を如何に科学的かつ効率的に評価するか、ということは、依然として各国の規制当局にとって重要な懸念要素となっている。日中韓3カ国の医薬品規制当局間の協力と臨床データ相互承認の促進により、企業はより柔軟な戦略的選択肢を得ることができるであろう。」
「中国の医薬品規制当局も日本の厚労省の措置から学び、中国がpivotal MRCTに参加する前に、中国の臨床データの要件をさらに明確に緩和し、より多くの良い新薬が早く承認されることを強く望んでいる」と丁锎博士は言った。
次いで、2番目のスピーカーである李皛氏は”How to Utilize Early Clinical Trial Data from China To Accelerate the Clinical Development of New Therapies in Japan”というタイトルで講演を行い、「日本だけで治験を実施する場合、患者登録が遅くかつコストが高いため、日中両国で同時に臨床開発を行う」ことを提案した。「具体的な臨床開発戦略には、ブリッジングや MRCT が含まれる。ブリッジングは、国間の開発時間差がある場合に適しており、先に開発を行った国からのデータは次開発国に外挿し申請資料として提出することが可能である。しかし、ブリッジング戦略ではドラッグラグが避けられず、複数国での同時開発をサポートできない。MRCT 戦略は多国で同時に開発し、同時に提出することができるため、最も効率的である。」

緑葉製薬(Luye Pharma Group)の国際研究開発臨床薬理学責任者である李皛氏
また、李皛氏は、「中国企業が国外で臨床開発を行う際、各地域の臨床開発チームが独自に開発戦略を立案し、臨床試験を実施し、多地域展開を行っているが、グローバル臨床開発チームでは一元的かつ統一的な管理と緊密な連携が必要で、計画と実行には、統一されたグローバル計画が必要である」と述べた。
日本での治験におけるファーマコビジランス関連規制

杭州泰格(Tigermed)の医薬品安全性副部長である呉翠翠氏
3番目のスピーカーは、CROである杭州泰格(Tigermed)の医薬品安全性副部長である呉翠翠(Wu, Cuicui)氏で、”What are the Special Requirements for Pharmacovigilance (PV) from Phase I to Phase III for PMDA in Japan?”とのタイトルにて、日本の治験におけるPV関連規制について紹介した。
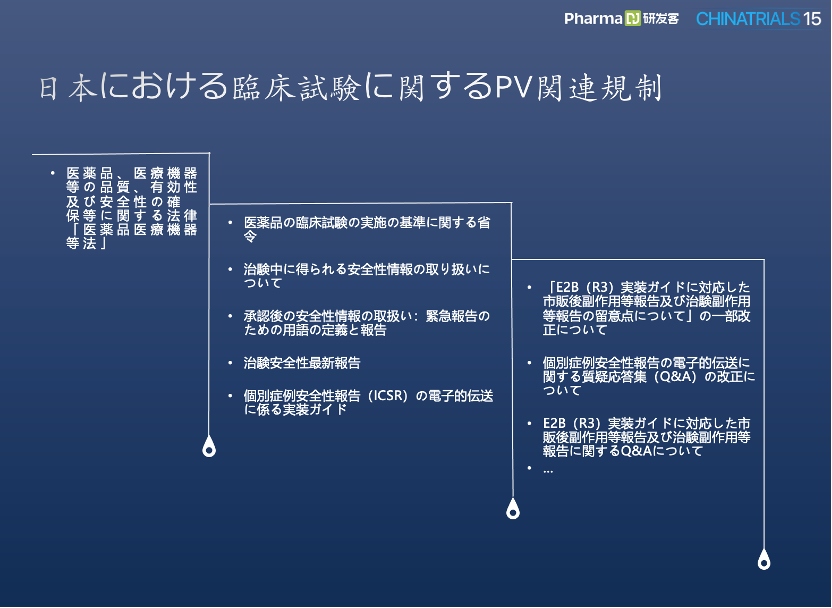
呉翠翠氏の講演スライドより
また、日本での治験開始前のPV関連業務の準備について、日本国内の現地PVベンダーの確認、安全性データベースの作成、安全管理計画書や報告書テンプレートの作成・レビュー、プロトコルの安全性報告書の関連章のレビュー、CRF (Case Report Form)、DMP (Data Management Plan)、CCI (Charlson Comorbidity Index) などのレビューなども詳細に説明した。最後に、中国の製薬企業が日本で治験を実施する際のSAE報告処理プロセスの事例も紹介した。
日本文化を理解し信頼を築く
セッション最後のパネルディスカッションには、上記3名のほか、日本で十数年間新薬臨床開発経験を持つ石薬集団(CSPC Pharma)チーフメディカルオフィサー項 安波(Xiang, Anbo)博士が加わった。同氏は、中国の製薬企業が日本で臨床開発を順調に遂行するためにはどのような事に注意すれば良いか、について述べた。

石薬集団(CSPC Pharma)チーフメディカルオフィサー項安波博士
治験運営に携わっている日本の大型病院は8000軒以上あり、治験のinvestigatorsは豊富な経験を有し、被験者の安全性を確保しながら、さまざまな創新薬の治験を高品質で完遂し、日本のCROも中国からの依頼を受け入れられようと積極的に体制を整えている。日本でも病院やinvestigatorsを選ぶプロセスは中国と似ており、治験を実施するに当たり、現地のinvestigatorsとの治験計画について意思の疎通を行う必要がある。日本のinvestigatorsもそれぞれ個性があって臨床開発に対して自分独自の見解を有する。日本のビジネス文化に馴染んでお互いの理解と信頼を築き上げることにより、プロジェクトの構築が容易になり、取引が成功する可能性が高くなる、と同氏は述べた。

本セッションの登壇者
パネルディスカッションの最後には、日本市場は米国、欧州に次いで注目すべき市場であり、日中共同臨床開発により効率向上とコスト削減が可能であり、中国企業の日本への展開は医薬品の成長に必要な過程である、と全員が一致した見解を示した。中国企業の日本における臨床開発が加速されるよう、日中両国の企業および規制当局が新たな道のりを共に進んで行くことを期待したい。
謝 辞:
日本語訳と編集、レビューをいただいた石薬集団チーフメディカルオフィサー項 安波(Xiang,Anbo)博士、東方伊諾の董 方(Dong,Fang)様、医薬品開発コンサルタント植村 昭夫博士、医薬研発達人編集長高野 哲臣氏に深く感謝申し上げます。
前号までの記事は下記からご覧いただけます。
第61号:DIA日本年会2023参加報告 ーChina Townhallを中心に
第60号:秦叔逵教授:「がんの王様-肝臓がん」の治療法の進展
第59号: 条件付き承認制度を厳格化する新たな規制案は中国の創薬企業に影響を与えるか?第58号: DIAアジア会議2023における中国の規制/業界関連のトピックス
第57号: CDE Annual Reports 2022を紐解く
第56号: 中国における自己免疫疾患治療薬の開発競争: 勝者は誰か?
第55号:CDEの『新薬のベネフィット•リスク評価』ならびに『患者を中心とする臨床試験のデザイン/実施とベネフィット•リスク評価』に関する技術ガイドライン
第54号: ASEANは中国のBiotech/Biopharmaにとって良い市場か?
第53号:中国のADC–10年の蓄積と3年の飛躍–
第52号:第15回DIA中国年会報告(第二弾) 医薬研発達人創刊2周年記念号
第51号:第15回DIA中国年会報告(第一弾) 中国バイオテク企業が日本へ進出するということ
第50号:呉洪福教授:中国は日本の幹細胞産業から何を学べるか
第48号:中国において抗悪性腫瘍薬の単群試験が適用となる6つのケース
第47号:2022 CBIICにて中国医薬品規制改革の成功の軌跡と今後の課題を見る
第46号:2023年度中国国家医療保険償還医薬品リスト(NRDL)交渉結果
第44号:中国における中枢神経系医薬品の研究開発:果てしない闇を越えて
第43号:中国の伝統的なジェネリック医薬品企業はどのようにして創新薬創出企業に生まれ変わったのか?
第42号:王娜アステラス中国開発本部長:アステラスは中国を含む世界同時開発を一歩一歩実現させる
第40号:中国は「患者を中心とする」新薬臨床開発の時代を開く
第39号:医薬研発達人第39号:2023年新春挨拶
第38号:25th CSCO 2022-CDE Session報告
第37号:海南博鰲(ボアオ)楽城国際医療観光先行区における「中国市場先行参入」の道
第35号:CDEが抗体薬物複合体(ADC)ガイドライン(案)を発出
第34号:DIA AsiaとDIA日本年会を通じて得られた中国に関する知見
第33号:住友ファーマ中国 纐纈 義隆氏:中国での新たな挑戦
第31号:中国における細胞及び遺伝子治療製品の審査概要と業界動向
第29号:中国における医薬品の臨床試験中ならびに市販後の変更
第28号:張 剣教授:中国の若手研究者により多くの成長の機会を!
第27号:中国バイオのイノベーションを投資市場の視点から切る
第26号:医薬研発達人創刊1周年に寄せて (2022年7月4日発行、第26号)
第23号:CDEのバイスペシフィック抗体医薬品ガイドラインがもたらすもの
第22号:中国製薬メタモルフォーゼ: BiotechからBiopharmaへ
第21号:中国における小児用医薬品開発の課題―現状を打破するにはー
第20号:2020年に登録された中国臨床試験の全体像から分かること
第19号:中国の製薬会社は、米FDAにおける信達生物製薬(Innovent)の経験から何を学ぶべきか?
第15号:2018~2021:過去4年間の創新薬の承認状況を振り返って
第13号:呉一龍教授:中国は世界の臨床研究において、重要な役割を果たしている
第12号:第6回中国医薬創新投資大会(CBIIC):導入(Buy)、フォロー(Follow)、改良(Improve)から真のイノベーションへ
第11号:CDE化薬臨床1部 楊志敏部長ご講演聴講記 (第18回DIA日本年会2021)
第10号:CDEが抗悪性腫瘍薬の臨床開発ガイドライン(案)を発出
第9号:2021年上半期の中国の製薬企業の導出・導入状況の分析
第8号: 中国GVP (Good Vigilance Practice) の公布・施行
第7号: 協和キリン丁 锎氏:日中両国臨床データ相互利用を強化する可能性
第6号:臨床的価値に焦点を当てる優先審査と特別審査 |上市促進プロセス(下)
第5号:中国の画期的治療薬、条件付き承認を多角的に分析|上市促進プロセス(上)
第4号:武田薬品の王 璘:中国の薬品研究開発:世界に追いつき、世界の研究開発をリードする
第3号:1回日中ICH合同シンポジウム:日中協働と相互理解の促進
第2号:NMPAはどのようにICH管理委員会メンバーに再選されたのか?
医薬研発達人の下記日本語ホームページにアクセスし、会員登録をしていただくと、自動的に記事の受信が可能となります。
Facebookアカウント:医薬研発達人、フォローをお願い申し上げます。
第62号

